

Understanding the Journey: A Parent’s Guide to DIPG
Edited by Ruth I. Homan, MPH
Copyright © 2012 by American Childhood Cancer Organization
®
(ACCO)
Printed in the United States of America
Published by American Childhood Cancer Organization
®
Cover and Interior Design: Ruth I. Homan MPH
Interior Layout: Ruth I. Homan, MPH, Devon Harp
Printing History: May 2012
ISBN 978-0-9854593-0-7
Library of Congress Control Number: 2012908135
e American Childhood Cancer Organization’s® name and logo are registered trademarks
of the national oce of the American Childhood Cancer Organization
®
. Visit us on the
web at: http://www.acco.org.
is book is written to provide information about childhood cancer and should not be
used as an alternative to receiving professional advice. Every eort has been made to ensure
that the information in this book is accurate at the time of printing, however, there is no
guarantee that the information will remain current over time. Always seek the advice of a
trained professional.
ACCO.ORG
®
Dedicated to all of the children who have battled or will battle
DIPG, and to Andrew Smith—for the impact you left on my life.
“It is only in the darkness, we can see the brightest stars.”
Martin Luther King Jr.
With the support of
Table of Contents
Preface vii
Part I. Understanding the Diagnosis
1. Brain Tumors 101 1
Violette Renard Recinos, MD, George I. Jallo, MD
2. Typical History of DIPG 17
Eric H. Raabe, MD, PhD, Kenneth Cohen, MD, MBA
3. Pontine Anatomy and Function 27
Sven Hochheimer, MD, Javad Nazarian, PhD, Suresh N. Magge, MD
4. Imaging DIPG 43
Jonathan Finlay, MD, Girish Dhall, MD, John Grimm, MD,
Stefan Bluml, PhD
Part II. Treatment
5. Clinical Trials for DIPG 71
Adam Cohen, MS, MD, Howard Colman, MD, PhD
6. Surgery: What it Can and Cannot Oer DIPG 89
Michael H. Handler, MD
7. Radiation erapy 101
Arthur Liu, MD, PhD
8. Radiosensitizers for DIPG 111
Roger J. Packer, MD
9. Chemotherapy and Biologics 123
David N. Korones, MD
10. e Use of Steroids in Patients with DIPG 139
Eric Bouet, MD, Ute Bartels, MD
Part III. Other Care Issues
11. Caring For Your Child At Home 153
Deborah Lafond, DNP, PNP-BC, CPON, CHPPN
12. Communication: When a Child Can No Longer Speak 193
Brownstone, MSW, RSW, Caelyn Kaise, MHSc, SLP (C), Reg. CASLPO,
Ceilidh Eaton Russell, CCLS, MSc (candidate)
Part IV. Research
13. Overcoming Research Hurdles in DIPG 223
Patricia Baxter, MD, Susan Blaney, MD
14. e Future of Genomics and Proteomics in DIPG 231
Mark W. Kieran, MD, PhD
15. Animal Models for DIPGs 239
Oren J. Becher, MD
16. Neural Stem Cells and DIPG 245
Michelle Monje, MD, PhD
17. Convection-Enhanced Delivery in DIPG 249
Zhiping Zhou, MD, PhD, Mark M. Souweidane, MD
18. Vaccine Treatment Strategies 269
Christopher Moertel, MD
Part V. End of Life Decisions
19. DIPG and Tissue Donation 273
Cynthia Hawkins, MD, PhD, Eric Bouet, MD, Ute Bartels, MD
20. Organ and Tissue Donation 285
Angela Punnett, MD, FRCPC
21. Integrating Palliative Care and Dicult Decisions 293
Justin N. Baker, MD, FAAP, FAAHPM, Adam J. Tyson, MD,
Javier R. Kane, MD
22. Journey of Sadness and Hopes: A Letter to Parents 319
Tammy I. Kang, MD, MSCE, Chris Feudtner, MD, PhD, MPH
Part VI. Appendices
A. Sample Medications Form 333
B. Glossary of Terms 335
C. Resources 343
D. Research Articles 355
VII
Preface
Preface
e American Childhood Cancer Organization’s (ACCO) mission is to provide
information and support for children and adolescents with cancer and their
families; to provide grassroots leadership through advocacy and awareness;
and to support research leading to a cure for all children diagnosed with this
life-threatening disease.
Since ACCO's founding in 1970, clinical research has increased the ve-year
survival rate of childhood cancer in the U.S. to approximately 80 percent. is
improvement in survival brings hope to tens of thousands of families whose
children are treated for cancer each year. In spite of the progress, however, too
many families still endure the loss of their precious son or daughter to cancer.
In the U.S. cancer continues to be the primary cause of death by disease in
childhood. As a result, families whose children are currently ghting this disease
need access to information to help them with the many treatment decisions
they must make; and there is an acute need for an increase in pediatric oncology
research funding that will lead to the development of new treatments for children
diagnosed with cancer in the future.
Among the many devastating childhood cancers, children who are diagnosed
with diuse intrinsic pontine glioma (DIPG) desperately need access to new
treatments. Regarded as the most aggressive of all pediatric brain tumors, all of
these children face a dismal prognosis. Currently, radiation therapy oers a short
reprieve from a ravaging disease. e tragedy of this disease is best expressed
in the following words from one parent's writing about her precious daughter.
"Imagine that you had a cherubic, mischievous, energetic and moody
two year old with ashing blue eyes, a brilliant smile and curly red
hair. Imagine that each morning she got you up at 5:15 a.m. by
standing up in her crib and shouting, "Maaamaaa, I'm awaaaake!
Maaamaaa, where are you?" Imagine if when you went into her
room she threw both her arms up towards you in a great big hug
and chattered her way into the living room, telling you she wanted
Cheerios for breakfast…with banana…and milk…and can we paint
now…and watch Caillou. Imagine if when you tried to get her dressed
Preface
VIII
IX
Preface
in the morning, she ran away from you laughing, no matter how
exasperated you got. Imagine if she insisted on picking out her own
clothes, and you let her, rather than ght about it. Imagine if she
could sing the entire theme song to "Golden Girls," could go down
the slide on her own, could pee on the potty, catch a ball, dance and
chase her friends. Imagine when you step o the subway after work
and walk into her daycare room, all the kids turn to look at who has
entered the room, and when she sees you she ashes the most brilliant
smile and comes running with her arms up, saying "Mama! Mama!
Mama!" Imagine if no matter how many times she had a tantrum
and demanded things from you and exhausted you, she ended each
night with a snuggle and a kiss and you breathed in the smell of her
curls and felt warm happiness all over. Imagine if you could never
love anything as much as you loved your rst born child, your dream
come true, your daughter.
Now imagine it is 9 months later. Imagine she is lying next to you in
your bed. She can't walk. She can't use her arms or hands. She can't
hold her head up. She can't see the television. She can't tell you she
loves you. She can't hug you. She is lying in the bed sound asleep,
but coughing on her own saliva, which she is starting to choke on
because she can barely swallow. Imagine she was dying and there was
nothing you could do to change it. Imagine if you knew that one
day soon you would never get to see her again. Never see her smile,
feel her hand slip into yours, kiss her warm cheek, feel her sigh into
your chest.
at is the simple reality of what we are living with. And it's hard. No
matter how many good things happen to us, no matter how much
we believe in a bright future for ourselves and a time of healing, we
are being tortured. No matter how well or easily we manage to get
through the days, to talk with our friends, to laugh and joke and
even ght sometimes, we are broken inside. It's a very strange way
to live. We need to not focus only on what we are losing, but on all
we have gained, but despair creeps in nonetheless.
What is keeping us moving forward right now, even when our hearts
are completely broken, is watching how our daughter has chosen to
live her short life. How she treats each day as a new adventure; pushes
herself both physically and mentally to ensure that she accomplishes
what she wants on that particular day. Sometimes it's something
big—painting with her mouth and visiting the pigs at the farm. And
sometimes it's just being able to mouth the words "ice cream," and
then napping most of the day. But she is always true to herself, and
even though things are hard for her, she ignores the barriers of DIPG
and chooses to forge her own path. Most importantly, she believes
that when life gives you a hundred reasons to cry, you need to nd a
thousand reasons to smile…And in my own smiles, I have become
familiar with the bittersweet taste of getting to parent my precious
daughter—the best experience in the world, but like a spring day
that is much, much, too short."
As this parent so eloquently states, having a child diagnosed with DIPG is the
most dicult journey that any parent will ever endure. is book was written to
help parents understand that journey so that they are better equipped to make
decisions regarding their child’s diagnosis, treatment, entry into clinical trials,
palliative care and quality of life during this critical time. It was also written to
provide hope for a future when children diagnosed with DIPG will be cured
of their disease and able to live long and healthy lives.
e Contributors
In November 2009, I was introduced to Andrew Smith—a magical young boy
who was battling DIPG. He was hospitalized at the National Institutes of Health
(NIH) in Bethesda, Maryland, where he and his family were making a decision
regarding his participation in a clinical trial. It was anksgiving, and through
Andrew's determination to communicate and the communication skills of his
parents with the "yes/no" questioning technique, we learned that the hospital
menu did not include pumpkin pie—a "must have" item during the holiday
season. We also learned that Andrew (who was a "foodie") would love not just
a piece of pumpkin pie, but an entire pie! I was blessed with making that for
Andrew, and also blessed with his life and the personal introduction to DIPG.
Around the same time, Loice Swisher—a friend and fellow childhood cancer
advocate—informed me of the need for a book that parents could turn to that
would assist them with their understanding of the disease and the treatment
decisions they needed to make on behalf of their children. We worked up a draft
of potential chapters and authors and thus began the "journey" of this book.
Our hope was to provide essential information from the diagnosis of DIPG
through to the end of life. Clinicians and scientists who were researching and/
or treating children with DIPG were asked to volunteer their time and expertise
Preface
X
XI
Preface
in the writing of this much needed resource. Every expert who was asked agreed
to participate—each contributing a chapter that detailed their DIPG research
and/or their clinical specialty.
To add a perspective from the 'trenches,' each chapter (with the exception of
the research chapters), is followed by personal stories written by parents. ese
sections entitled “Parent Perspectives” illustrate the despair and the hope that
accompany the DIPG experience. As with all hardships, those individuals
who have endured life's burdens become experts in their personal journeys.
Like the professionals who are researching and treating our nation's children
with brainstem glioma, parents of children with DIPG also became experts in
knowing how to provide the best of care for their children.
I am deeply indebted to the following professionals who gave of their time and
expertise to author comprehensive chapters for this book. My deepest gratitude
is extended to: Violette Renard Recinos, MD, George I. Jallo, MD, Eric H.
Raabe, MD, PhD, Kenneth Cohen, MD, MBA, Sven Hochheimer, MD, Javad
Nazarian, PhD, Suresh N. Magge, MD, Jonathan Finlay, MD, Girish Dhall,
MD, John Grimm, MD, Stefan Bluml, PhD, Adam Cohen, MS, MD, Howard
Colman, MD, PhD, Michael H. Handler, MD, Arthur Liu, MD, PhD, Roger
J. Packer, MD, David N. Korones, MD, Eric Bouet, MD, Ute Bartels, MD,
Deborah Lafond, DNP, PNP-BC, CPON, CHPPN, David Brownstone, MSW,
RSW, Caelyn Kaise, MHSc, SLP (C), Reg. CASLPO, Ceilidh Eaton Russell,
CCLS, MSc (candidate), Patricia Baxter, MD, Susan Blaney, MD, Mark W.
Kieran, MD, PhD, Oren J. Becher, MD, Michelle Monje, MD, PhD, Zhiping
Zhou, MD, PhD, Mark M. Souweidane, MD, Christopher Moertel, MD,
Cynthia Hawkins, MD, PhD, Angela Punnett, MD, FRCPC, Justin N. Baker,
MD, FAAP, AAHPM, Adam J. Tyson, MD, Javier R. Kane, MD, Tammy I.
Kang, MD, MSCE, and Chris Feudtner, MD, PhD, MPH.
My thanks would not be complete without expressing my deepest appreciation
to all of the parents who took their time to write and share their children's
personal stories. ere are 164 writings by parents in this book. ese stories give
credibility and passion to the book and emphasize why the book is so critically
needed. ese stories also serve as a reminder that these are the precious lives of
children who just want to grow up.
e words "ank you," cannot adequately express my gratitude for the time
that Sandy Smith spent pulling together the parent stories. Sandy is a woman
who has turned her own grief, from losing her son to DIPG, into generously
giving of herself to help DIPG families navigate their journeys. She makes herself
available—whether it's to assist a family as they are starting down this path, or to
facilitate tissue donation at the end of life. She makes herself available as a trusted
patient navigator and friend. As a result of working together on this book, I too
am proud to be able to call Sandy a very dear friend. I also want to personally
thank Kim Spady, Jonathan Agin and Nettie Boivin for the time they spent
reviewing parent stories, providing personal insight into the DIPG journey, and
for answering my emails day or night during the nal production phase of this
book. Your generosity of time and wisdom are appreciated more than you will
ever know.
My heartfelt appreciation is extended to Dr. Andrew von Eschenbach for the
thoughtful and compassionate words that he provided for the back cover copy
of this book. I am forever grateful for his commitment to childhood cancer
research and the role that he played during his term as Director of the NCI to
provide funding for TARGET—an initiative that utilizes genomic technologies
to identify therapeutic targets in childhood cancers (http://target.cancer.gov).
His belief that increasing eorts to target and control cancer by modulating and
altering the behavior of the disease on a molecular level directly resulted in the
funding of this innovative childhood cancer research project.
I am personally grateful to Dr. Peter Adamson, Chair of the Children's Oncology
Group, for also contributing to the back cover copy text of this book. Dr.
Adamson's dedication to children with cancer as a clinician, and his strong
leadership as chair of the world's largest pediatric oncology clinical trial research
group, is inspirational.
Finally, I wish to thank Ryan and Maria Reilly for sharing the picture of their
precious son Liam which graces the cover of this book. Liam responded to
radiation and was given the "gift of a honeymoon period." His "bucket list" during
that brief time included visiting the special place where this photo was taken.
By sharing Liam in this way, Ryan and Maria have personalized the disease for
many, and are building awareness of DIPG as a result. anks as well to Marie-
Dominique Verdier of Sand Point Photography for her help with enhancing and
cropping the photo for the book cover. My sincere gratitude is also extended to
ACCO board member Nicole Roman for suggesting the title of the book as a
result of her daughter Sophia's childhood cancer journey.
Book Composition
is book is written for both parents of children diagnosed with DIPG, as well
as health care providers. Chapters are written independently so parents and
providers are encouraged to read those chapters that most directly apply to a
Preface
XII
XIII
Preface
child's current medical needs. For example, parents with a newly diagnosed
child might want to start with chapter 10: e Use of Steroids in Patients with
DIPG. Others might want to read the book from start to nish.
e book is divided into six major sections. Part I: Understanding the
Diagnosis provides an overview of diuse intrinsic pontine glioma including
the diagnosis and typical history, pontine anatomy and function, and DIPG
imaging.
Part II: Treatment outlines treatment related issues including clinical trial
design, surgery, radiation, radiosensitizers to treat DIPG, chemotherapy and
biologics, as well as the use of steroids.
Part III: Other Care Issues identies additional treatment concerns including
caring for your child at home, and provides information to assist with
communication when a child can no longer speak.
Part IV: Research provides an overview of the hurdles to DIPG research, as
well as the hope found through genomic and proteomic techniques, animal
model research, neural stem cell research, convection-enhanced delivery, and
vaccine treatment strategies for DIPG.
Part V: End of Life Decisions addresses the dicult questions that families
face as they come to the end of life stage. ese include autopsy tissue donation,
organ and tissue donation, as well as integrating palliative care while making
dicult decisions. e book concludes with a letter of hope written by two
physicians to families of children with DIPG.
Finally, there are four useful appendices at the end of the book.
• Appendix A includes a sample medications form.
• Appendix B provides a glossary of medical and research terms used
throughout the book.
• Appendix C lists a sampling of resources—books, websites, listservs, and
organizations that assist families of children with DIPG.
• Appendix D is a compilation of journal articles that provides further
reading opportunities for those wishing to dig deeper into a specic topic.
Acknowledgements
is book would not have been possible without nancial support. I am indebted
to Mr. and Mrs. A. James Clark, and Courtney Clark Pastrick of the Clark
Charitable Foundation for their generous donation that made the printing of this
book possible. eir commitment to making a dierence in the lives of children
is inspirational. ey are true heroes to children with cancer.
My heartfelt thanks to Joseph E. Robert, Jr. and e Team Julian Foundation,
founded by his family in loving memory of courageous Julian B. Boivin, for their
nancial support to cover additional expenses related to the distribution of the
book to families.
Finally, I am deeply grateful to Christine Martin for her nancial support from the
Just One More Day Foundation, which she started to honor the memory of her
daughter Alicia. rough her generosity, parents of children diagnosed with DIPG
will have access to information that will provide them with an understanding of
this dicult journey and assist them with the decisions they must make.
I come away from this book with a renewed burden for our nation's children
with cancer and their families. It is not good enough that 80 percent of America's
children with cancer survive. It is not acceptable that children diagnosed with
DIPG don't grow up to live out their dreams. We must never tire of the need
to increase awareness of the impact of childhood cancer in the United States.
As parents of children with cancer, and their advocates, we must continue to let
our voices be heard. We must continue to knock on politicians' doors and insist
that research for childhood cancer receive priority funding. We must continue
to do our part to raise funds to build programs that help children being treated
for cancer today, as well as research funds that will help children diagnosed with
cancer tomorrow.We must continue to work together in the belief that doing so
is the only way to make a dierence, and doing less is simply not good enough.
Ruth I. Homan, MPH
Executive Director, Editor
American Childhood Cancer Organization
Preface
XIV
“If there ever comes a day when we can’t be together, keep me
in your heart, I’ll stay there forever.”
Winnie the Pooh

1
Chapter 1: Brain Tumors 101
Chapter 1
Brain Tumors 101
Violette Renard Recinos, MD
George I. Jallo, MD
Brain tumors are the most common solid tumor found in the pediatric
population. Each year, approximately 3,400 children are diagnosed with a
primary tumor of the central nervous system (CNS)—comprising the brain and
spinal cord. While all primary brain tumors arise from cells originating in the
brain, these CNS tumors can dier signicantly with regards to location, cell
origin and pathology, clinical manifestations, prognosis, and treatment options.
Some of these tumors may exhibit a slow benign (noncancerous) growth pattern,
while others are more aggressive and classied as malignant or cancerous. Due
to the sensitive, important structures of the CNS, even a benign tumor may
cause signicant clinical symptoms if located in or near critical brain or spine
structures. Similarly, surgical access to these so-called “benign” brain tumors can
be limited or dangerous, making the tumor inoperable and thus the potential
to act as a more malignant lesion over time.
Advances in imaging with computed tomography (CT) and magnetic resolution
imaging (MRI) allow clinicians to better visualize the mass and determine
key characteristics that may help dierentiate one tumor from another. By
evaluating the tumor size and shape, identifying its location and eect on
adjacent structures, and examining patterns of contrast enhancement (a
substance that enhances the contrast of body structures on medical imaging),
clinicians may be able to diagnose the tumor with imaging alone. Sometimes
further studies, such as blood work or cerebrospinal uid (CSF) analysis by
lumbar puncture—also called a spinal tap—can also help with the diagnosis.
When feasible, direct histological evaluation by either biopsy or tumor resection
is the gold standard in diagnosing the specic tumor type. Often, however,
the imaging, together with clinical presentation and a laboratory workup, can
help clinicians formulate the best suitable
treatment option for the patient.
Treatment of brain tumors can vary widely
Dr. Recinos is a Pediatric
Neurosurgeon at the Cleveland
Clinic Foundation, Cleveland, OH.

Chapter 1: Brain Tumors 101
2
3
Chapter 1: Brain Tumors 101
and may include close clinical observation with interval imaging, surgical
resection, chemotherapy, radiation, or a combination of these modalities. e
exact combination of therapies will depend upon the expected behavior of
the tumor, and clinicians will need to carefully weigh the risks and benets of
the treatments. Pediatric tumors and their treatment dier from their adult
counterparts, especially as long-term eects from chemotherapy and radiation
have a greater impact on the developing CNS of a child.
Epidemiology of Brain Tumors
Brain tumors are the leading cause of cancer death in the pediatric population.
ey are the second most common malignancy in children behind leukemia, and
the most common solid tumor found in the pediatric population. According to
the 2009 statistical report of the Central Brain Tumor Registry of the United
States, 7% of all reported brain tumors are found in patients younger than
20 years of age. e overall incidence of brain tumors found in the age group
comprising 0–19 year olds is 4.58 per 100,000 individuals. Overall, there is a
slight male predominance in pediatric brain tumors, however, depending on
the individual histology, certain tumor subtypes are found more frequently in
females. Brain tumors are also more common in whites than blacks. e age
distribution varies depending upon the specic tumor, with certain tumors
such as pilocytic astrocytomas, malignant gliomas, and medulloblastomas more
common in the younger pediatric population of 0–14 year olds, while germ
cell tumors are more common in 15–19 year old age group.
e majority of pediatric tumors are located in the cerebral hemispheres, mainly
within the frontal, parietal, temporal, or occipital lobes, overall making up 24%
of brain tumors. Sixteen percent of tumors are found in the cerebellum, 12%
in the brainstem, 6% within the ventricles, 11.6% in the pituitary, and 3.2%
in the pineal region. e remaining less common sites include the meninges,
the cranial nerves, the spinal cord, and other brain areas.
Brain Tumor Histology and Classication
ere are more than 120 dierent classications of tumors, most of which are
classied by the cell type from which they arise. A better understanding of this
classication system can be gained through
a brief review of the cells that comprise the
central nervous system and its coverings.
As a very general overview, the majority
of CNS cells can be divided into two groups: neurons, which are cells that
send and receive electrochemical stimulation to and from the brain and spinal
cord, and glial cells, the cells that support neurons. ere are many dierent
types of neurons found throughout the brain which carry a variety of signals
depending on their function and location. Unlike many other cells in the body,
neurons, do not regenerate after damage, although there have been notable
exceptions to this rule. Glial cells are far more plentiful than neurons, making
up about 90% of brain cells. ese cells are further specialized to provide specic
functions to support the neuronal tissue. Some glial cells function to provide
structural or nutritional support, while others help to insulate the neurons,
provide defense against pathogens, and clean up cellular debris. e most
common glial cells are astrocytes, oligodendrocytes, Schwann cells, microglia,
and ependymal cells. Covering the brain and spinal cord is a membrane called
the meninges, comprised of meningothelial cells. Tumors may arise from any
of these subtypes and thus be named according to their cells of origin. Hence,
the terms astrocytoma, oligodendroglioma, ependymoma, etc. may sound
familiar. You may frequently hear of tumors referred to as gliomas, or glial
tumors. ese are generally referring to tumors arising from astrocytes as these
are most commonly the cell of origin.
e World Health Organization (WHO) has categorized gliomas into four
classications based on tumor aggressiveness and malignancy. WHO grade I
tumors are low-grade lesions that are non-inltrating and have well-dened
borders. ese tumors can be cured if in a location amenable to surgical
resection. ey are slow growing and may remain inactive, even if they are
not completely excised. Examples of WHO grade I tumors include pilocytic
astrocytomas and pleomorphic xanthoastrocytomas. WHO grade II tumors are
more inltrative. ese tumors tend to invade normal tissue, making complete
surgical resection more challenging than WHO grade I lesions, especially if
the tumors are located in critical brain structures. ese tumors tend to be
slower growing than the higher-grade lesions, but they may evolve into WHO
grade III or IV lesions over time. Examples of WHO grade II lesions are
oligodendrogliomas and low-grade astrocytomas. WHO grade III tumors are
known as anaplastic astrocytomas. ey are considered malignant because they
are inltrating, fast growing, and often require treatment with surgical resection,
chemotherapy, and radiation. Grade IV lesions, also known as glioblastoma
multiforme (GBM), are even more aggressive than anaplastic astrocytomas and
may grow so rapidly that they outgrow their blood supply and cause tumor
necrosis. GBMs are malignant lesions with poor prognosis regardless of the
treatment (e.g., surgery, radiation, and chemotherapy).
Dr. Jallo is a Professor of
Neurosurgery, Oncology, and
Pediatrics at the Johns Hopkins
Children’s Center, Baltimore, MD.

Chapter 1: Brain Tumors 101
4
5
Chapter 1: Brain Tumors 101
Another cell type to note is the “embryonal” cell. In development, the neurons
and glial cells are thought to derive from a common progenitor cell that
dierentiates at dierent stages depending on certain genetic signaling and
other local factors. Several tumors in the pediatric population have features
that resemble this more primitive “embryonal” cell. ese tumors can be very
undierentiated or they can contain certain features that place them into a
more specic category depending on cytoarchitecture or immunohistochemical
staining. Included in this group are tumors such as medulloblastomas, primitive
neuroectodermal tumors (PNET), and atypical teratoid/rhabdoid tumors (ATRT).
Germ cell tumors are another group of tumors requiring special note. As with
embryonic tumors, germ cell tumors arise from cells that are not commonly
found in the adult brain. ese tumors originate from cells in the developing
embryo’s yolk sac endoderm that migrate throughout the embryo. ey
frequently are found in the pineal and suprasellar/pituitary region, as well as
the third ventricle and posterior fossa. Germinomas are the most common germ
cell tumors in the pediatric population.
Classication of Brainstem Gliomas
Overall, the most common brain tumors found in the pediatric population are
pilocytic astrocytomas, malignant gliomas, and medulloblastomas. Within the
glioma category, brainstem gliomas constitute 10–20% of all pediatric CNS
tumors. We will narrow the scope of our discussion to brainstem gliomas, as
they tend to present a unique and challenging tumor with its own classication
scheme that provides a framework to predict growth patterns, surgical
resectability, and overall prognosis.
Many classication schemes have been devised to categorize brainstem tumors
based on imaging and tumor characteristics. All of these systems categorize
tumors based on diffuse or focal imaging characteristics. Several more
complex classication systems further divide the tumors based on location,
growth pattern, and presence of hydrocephalus or hemorrhage. All of these
characteristics can be determined with a high-quality MRI image [Table 1].
1986 Diuse
Focal
Circumscribed mass less than 2 cm, no edema
Cervicomedullary
1991 Location
Midbrain, pons, medulla
Focality
Diuse or focal
Direction and extent of tumor growth
Degree of brainstem enlargement
Exophytic growth
Hemorrhage or necrosis
Evidence of hydrocephalus
1996 Focal
Midbrain, pons (dorsal exophytic pontine glioma), medulla
Diuse
Table 1: Classication Schemes for Brainstem Tumors
Figure 1a: Sagittal T1-weighted MRI showing the diuse pontine glioma with expansion
of the pons.

Chapter 1: Brain Tumors 101
6
7
Chapter 1: Brain Tumors 101
One of the main characteristics determined on imaging is the degree of focality;
in other words, is the tumor diuse and inltrating or does it have a clearer
demarcation of margin? Diuse gliomas make up 58–75% of all brainstem
tumors and are the most common tumor found in this location. On MRI
imaging they have indistinct margins and
are characterized by diuse inltration
and swelling of the brainstem [Fig.
1 a, b, c]. ese tumors are usually located
within the pons, however they may also extend into other areas of the brainstem.
ese tumors have variable contrast enhancement and tend to be high-grade
lesions. e usual histopathology is typically a malignant brillary astrocytoma,
WHO grade III or IV.
Figure 1b: Axial Flair MRI sequence showing the pontine glioma, which involves the
entire pons.
Focal tumors have more clearly dened margins and, when in the brainstem,
are usually found in the midbrain, pons, or medulla [Fig. 2]. ey usually
are not inltrating and are not associated with swelling of adjacent structures,
also known as edema. ese focal tumors are often benign on histology and
Figure 1c: e axial T1 image shows the noncontrast enhancing and inltrative tumor.
graded as WHO grade I or II, although cases of more aggressive tumors have
been reported.
In addition to degree of focality, some classication schemes also consider
whether the tumor is primarily inside the brainstem, which is dened as
intrinsic, or if it resides mostly outside the brainstem, which is dened as
exophytic. Exophytic brainstem gliomas arise from the subependymal glial
tissue and the majority of the tumor is located in the fourth ventricle. ese
are usually well-dened tumors that are almost always low-grade gliomas.
Location of brainstem tumors is also a consideration when classifying them.For
example, cervicomedullary tumors, found where the lower part of the brainstem
connects to the top of the cervical spinal cord, tend to be slow growing and
focal lesions and thus are considered benign low-grade astrocytomas. However,
more aggressive cervicomedullary tumors, which are more inltrative and grow
up into the brainstem, have been found in this location.

Chapter 1: Brain Tumors 101
8
9
Chapter 1: Brain Tumors 101
Figure 2a
Figure 2b
Using these classication schemes—combining degree of focality (i.e. diuse
or well delineated), intrinsic or exophytic, and tumor location—health care
providers can formulate a dierential diagnosis and establish a reasonable
treatment plan. Characteristics that may help further classify the tumor include
direction and extent of tumor growth, degree of brainstem enlargement,
hemorrhage or necrosis, and evidence of hydrocephalus.
Another simpler classication scheme divides brainstem tumors into typical and
atypical brainstem gliomas. e term typical brainstem gliomas is synonymous
with the term diuse intrinsic pontine gliomas (DIPG). As mentioned earlier,
these tumors are diuse and inltrative, located in the pons but potentially
extending into other areas of the brainstem. Surgery or biopsy of these lesions
is not usually recommended at this time, unless the diagnosis is in question.
Figure 2c
Figure 2a: Sagittal T1-weighted MRI with contrast shows a focal enhancing tumor in the
pons and midbrain. Figure 2b: T2 weighted axial image shows the focal noninltrative
or diuse nature of the tumor. Figure 2c: T1 weighted axial image shows a focal tumor,
which is not invading the pons or brainstem; this is a benign juvenile pilocytic astrocytoma.
Chapter 1: Brain Tumors 101
10
11
Chapter 1: Brain Tumors 101
Parent Perspectives
Our world splintered into millions of pieces on Sunday, February 1st, when
Bryce at age 13 was sitting in church with us and he turned to look at me.
His eye turned in toward the center. Then it went back. I almost wasn’t
sure that I had seen it happen. Then, later in the day, Bryce had a hockey
game, and came off the ice to tell us that he could see two hockey nets, not
just one, and that “he had shot at the wrong one and missed.” We knew
something was wrong, and on Monday, we called for an appointment with
our family doctor. He had us in to see an eye specialist by that afternoon.
On February 10th, Bryce had a CAT scan, which didn’t show anything.
Luckily, the specialist was persistent, and he ordered an MRI. He told us
that at 13, an eye turning in was not “normal” if he didn’t have it at birth.
And so, Bryce’s journey began. On Feb. 26th, Bryce went for an MRI
where we were whisked out of the waiting room by a radiologist and sent
to Bryce’s pediatrician, who told us about the growth—a diffuse intrinsic
pontine glioma, which is a tumor located completely inside the brainstem.
He told us that treatment would involve going to Children’s Hospital, and
that he wanted us there that night. We were floored. I don’t remember much,
but I remember looking at Bryce’s face, and his eyes filling with tears. He
didn’t say much. None of us did, I think we were in shock. We went home
and packed a bag, and of course, family started pouring into our house
as we were preparing to leave. I was crying, and he just looked at me and
said, “It’s just cancer, mom. It will be fine.”
Blood was drawn and another series of neurological tests were performed.
Ellie was incredibly brave for the scan and held very still as the machine
whirled around her head. I remember being outside her room trying to find
water and hearing the emergency room doctor being paged by a doctor we
know through the girls’ school. My stomach jumped to my throat. An IV
was administered and an MRI was ordered. Ellie was very scared as she
felt fine and did not understand what was going on and why she needed
all these tests. She was exhausted.
My husband stayed with Ellie while my father, Ellie’s pediatrician and I
Atypical gliomas include the focal lesions which are well circumscribed. ey
may be contained in the brainstem or may grow out in cysts or outside of the
brainstem. Unlike typical brainstem gliomas, the atypical gliomas tend to arise
from the midbrain (the top of the brainstem) or the medulla. ese atypical
tumors tend to be lower grade lesions which may be amenable to some degree
of surgical resection or biopsy.
e majority of brainstem tumors are diuse pontine gliomas that are mainly
high grade on histological examination and have poor prognosis. In this book,
we will review the current treatment strategies, role for surgery, radiation and
chemotherapy, and future directions in the treatment of this disease.
Chapter 1: Brain Tumors 101
12
13
Chapter 1: Brain Tumors 101
met with the neurosurgeon in a private area to view the scan. I really had
no clue what I was looking at but a child’s head. I looked around at the
three doctors and watched my father and our pediatrician find chairs. Their
faces said it all. Ellie’s golf ball size inoperable tumor was situated in the
pons of her brain stem and had grown to the point where it was sitting on
nerves that obviously affected her left side.
We spent the next three nights in the hospital medicating, testing and
meeting an army of staff to try and assist us in navigating through this
whole new world. We heard the words diffuse intrinsic pontine glioma…
can you please write that out? We were told surgery was not an option nor
was biopsy. We were devastated and heartbroken and my husband and I
challenged each other every moment of every day to keep our spirits high,
in order to be strong for Ellie.
The diagnosis came quick after a trip to Children's Hospital’s emergency
room. We were in a complete haze. I knew I needed to get a second opinion
and didn't know how to go about it and unbelievably I felt sheepish about
asking for the copies of the results—even though I did.
The second opinions came slow but we knew we had to act fast for any
treatment. We weren't given much hope but were told that radiation would
be the only thing that would relieve her symptoms and buy her some time;
maybe a year or two.
There was no chemo that anyone had any confidence in but we could always
try one that is well tolerated by some other kids. We did the exhausting
process of radiation and they said it would get worse before it gets better
and it did. After the radiation we thought we saw signs of the honeymoon
period. Unfortunately, the honeymoon was held off by an infection of her
shunt. After that was cleared we saw a fast improvement and we finally had
our old Tatumn back! But that time was soooo quick. By the time the steroids
were totally out of her system and her hair started to grow back from the
radiation her symptoms subtly started to reappear. Then her journey ended
very quickly. It was about 5 1/2 months total from diagnosis.
Lovis first had a CT scan at our local hospital after which we were told
about a mass in her cerebellum, most likely to be operable. Because it
was a Saturday night, we had to wait to be transferred to the Children's
Hospital where an MRI was done on Sunday night. The wait for the results
was terrible. Lovis was completely exhausted, weak and almost could not
speak or drink.
On Monday morning at 10:00 a.m. we were shown the scans and were
told that Lovis had diffuse intrinsic pontine glioma that presents itself as
tiny little spots all over the pons in the brainstem—thus inoperable and
sentencing our daughter to death in about 9 months even if we chose to
radiate. We fell apart. I remember screaming "No, no" and then turning
back to the screen, wanting to know EVERYTHING about this tumor. So
one nurse left and printed off more information right away.
We immediately opted for radiation, both my husband and I. But Lovis was
too weak for the daily sedation that goes with it since she had just turned
three. We were told to wait, try steroids and temozolomide for lack of better
agents, aware that the latter would not be a great help. The doctors thought
that even one general anesthetic would have been enough to kill Lovis if
we had started to radiate right away. If she didn't get better within a few
days, stronger and able to be sedated, she might likely die within the next
10 days. That was the diagnosis.
I was diagnosed with breast cancer on a Monday afternoon. Three days later
we ended up in the emergency room with my 6 year-old-son Andrew. After
a CT scan, a young physician's assistant came to talk with us, and I will
never forget her words. "There is a large area of swelling in the brainstem.
We suspect a mass." Andrew responded casually, "My mom has a mass!"
When I think of a mass, I think of a ball or an egg or something that can be
removed. I remember being told that there were parts of the mass reaching
out like fingers into the brain. It was not until eight months after diagnosis
that I truly understood the meaning of the word diffuse. We were not dealing
with fingers reaching into the brain. We were dealing with cancer cells
sprinkled among healthy brain cells in the pons (part of the brainstem).
Andrew's neuro-oncologist explained it by using the idea of sand (cancer
cells) in grass (healthy brain tissue). I have also heard people use the idea
of marbling in steak.
Chapter 1: Brain Tumors 101
14
15
Chapter 1: Brain Tumors 101
Our oncologist and neurosurgeons we consulted with, told us the hard
truth but to the point that my husband and I were so discouraged that
we almost did nothing. Because of Connor's age they did not recommend
radiation and the evidence of effective chemo was null. We almost felt as
if we were dismissed with all of our questions and concerns because of the
known outcome with this type of tumor. We did go ahead with the radiation
and Connor was great for several months until the tumor didn't follow the
"typical course" so we were then sent for a third opinion and subsequently
a biopsy.
Six months after the original diagnosis we found out that Connor's pathology
report came back with a different tumor type. He had a new diagnosis of
a PNET which was some positive news we thought. The only problem was
by the time we found this out it had progressed in the brain and into the
spinal cord.
We did not know until a few months after diagnosis that a pediatric
oncologist is not a specialist in pediatric brain tumors. We were very
happy with our son's medical care, but we wish we would have understood
earlier the importance of having a pediatric neuro-oncologist involved
in the situation. Our level of understanding about our son's brain tumor,
possible treatments and related issues changed drastically as soon as we
established a relationship with a pediatric neuro-oncologist. Even though
this specialist was located in another state, she worked well with our son's
local team to manage his medical care. Looking back, that was the best
decision we made.
My mom, Bizzie and I headed to the emergency room. When we got there
we were taken into a room and told we were going to have to get Bizzie to
lie still for a CT scan. Well, when they talked about possibly sedating her,
I called my husband. Funny, but back then I could not even think about
holding her while she was sedated or undergoing a procedure. That would
change. I did get her to stay still for the CT scan by singing Row, Row, Row
your Boat to her repeatedly. I remember looking into the booth and seeing
the emergency room doctor look at the results. The technician walked us
back to the emergency room. They all knew. We did not—yet. My husband
was there when I returned. The emergency room doctor came in and spoke
to us, “There is a mass on her brainstem.”
I ran out of the room hysterical; but I returned in a minute. I needed to hear
this and be there for Bizzie. I remember snippets. She needed an MRI, which
meant an IV and sedation. The doctor told us that he suspected that it was
a diffuse intrinsic pontine glioma. The MRI would confirm this. She also
needed surgery to drain the fluid around her brain caused by the mass. We
would be admitted and she would be started on steroids. My husband asked
on a scale of 1 to 10 how bad was this? The doc quickly replied, “It’s bad.”
So Bizzie had her MRI. The anesthesiologist was impressed by Bizzie’s
strength when she came to. “Wow, she would do great in a bar-fight.” She
was kicking and hitting. I finally got her to calm down and we settled into
room 4 of the PICU. The pediatric neurosurgeon was sent the images from
the MRI and came in to talk to us about them. I sent my husband out to
talk to her. I stayed with Bizzie. When they came back, he had his poker
face on. He explained to me that it was a big tumor, and it could not be
removed surgically.
At that point I cut him off and turned to the neurosurgeon. "So, it’s
inoperable. What now?” She explained that standard treatment was
radiation followed by chemo, but that chemo was proven ineffective with
these types of tumors. I knew what she was saying. “So, how long?” I asked.
“Six to twenty-four months,” was her reply.
At that point Bizzie pulled my face back towards the book we were reading.
“Momma…READ!” I kissed her sweet little head. “Of course Bizzie, how
silly of me to not be paying attention to you.”
And that was diagnosis day.
At 3:00 p.m. I received a call from Liam's pediatrician's office. They asked
that we come in and that it would be better if we could leave our children
with a friend. I begged the woman who called to please tell me what was
wrong over the phone. She was very kind, but of course could not. They told
us we needed to come as soon as possible. I called my husband who was
at work a half hour away. I sat on my bathroom floor with the door locked
barely able to tell him he needed to come home, that something was wrong
with Liam's scan. It was the longest 30 minutes of my life while I waited
for him on our front porch. We drove in silence and my heart pounded. We
Chapter 1: Brain Tumors 101
16
sat for only moments in the doctor’s waiting room. They brought us into
an exam room where Liam's pediatrician gave us the news that they found
a lesion on his brain. I looked right at this woman and every part of me
thought she had to be to be lying to us and I just couldn't understand why.
Before I even realized, I heard myself calling this poor woman a liar and
asking her why she would lie about our son like that. She quickly came and
took my hand. I apologized after a moment. With great compassion she told
us what needed to happen next and that those plans had already been set in
motion. We would take Liam to the University Hospital the next day. Our
appointment was with a pediatric neuro- oncologist. In those very moments,
everything became marked with a new definition of time. Everything was
now defined as "before" and "after."

17
Chapter 2: Typical History of DIPG
Chapter 2
Typical History of DIPG
Eric H. Raabe, MD, PhD
Kenneth Cohen, MD, MBA
e brainstem is divided into three major areas—midbrain, pons and medulla—
and dierent types of tumors can occur in any of these locations. A glioma is a
tumor that arises from a cell in the brain called a glial cell. One type of glioma
is called an astrocytoma, which is a tumor that arises from a specic type of
glial cell called an astrocyte. e two major types of astrocytomas are pilocytic
astrocytomas and diuse, inltrating astrocytomas. e term DIPG specically
refers to a diuse, inltrating astrocytoma that develops in the pons.
Who is Aected by DIPG?
Each year approximately 200 children in the United States are diagnosed with
DIPG.e age range is broad, but the most common age at diagnosis is 7 to 9
years. All races and both sexes are equally aected.
Why do certain children get DIPG?
In short, doctors don’t know why. ere are no known associations of DIPG
with any environmental or infectious agents. Most researchers who study
DIPG believe these brain tumors, similar to other tumors aecting children,
arise when normal developmental and maturational processes go awry. In
this case, developing brain cells accumulate alterations in their DNA that
prevent them from properly maturing. ese alterations allow the developing
brain cells to continue growing, and this growth eventually becomes out of
control, leading to cancer. During the process of uncontrolled growth, DIPG
cells can gain DNA alterations that allow
them to resist the eects of radiation and
chemotherapy, making these cancer cells
extremely dicult to kill.
Dr. Raabe is an Instructor
in the Divison of Pediatric
Oncology, and Physician-
Scientist at the Sidney Kimmel
Comprehensive Cancer Center
at Johns Hopkins, Baltimore, MD.

Chapter 2: Typical History of DIPG
18
19
Chapter 2: Typical History of DIPG
Clinical signs of DIPG
e signs and symptoms of DIPG can start gradually. As described in chapter 3,
the pons contains nerve centers that control eye movements, facial movements,
swallowing, and speech. As pontine glioma tumors grow, the cancer cells
interfere with these centers, causing disruption of their functions. Sometimes
parents notice odd eye movements, slurred speech, diculty swallowing, and
trouble maintaining balance, or drooping of one part of their child’s face. e
pons also contains nerves that run from the brain to the rest of the body. Pontine
tumors can press on and interfere with the function of these nerves, leading to
weakness in an arm and/or a leg.
Tumors in the brainstem can also cause increased pressure within the skull.
e swelling from the tumor can cause increased pressure directly, or it can
block the ow of spinal uid from the skull (where it is made) to the spinal
cord (where it is absorbed). Increased pressure can cause patients to complain
of persistent headaches and in some patients can lead to nausea and vomiting.
ese daily signs of increased pressure inside the skull will get worse over time,
as the tumor grows.
DIPG Appearance on MRI
A magnetic resonance imaging (MRI) scan is the best non-invasive way to
determine the size and properties of brain tumors. DIPGs have a characteristic
appearance on an MRI, which other tumors that grow in the pons or other
parts of the brainstem do not share. e boundaries of a DIPG are dicult to
determine, because the tumor cells invade the surrounding tissue of the pons.
A DIPG generally does not have portions of the tumor that push outside of
the pons’ normal structure. In contrast, a pilocytic astrocytoma, another less-
aggressive brainstem tumor, has a more focal appearance, is more likely to have
a part that buds out of the normal structure of the brainstem, and will displace
rather than invade surrounding brain tissue. e dierences between how a
pilocytic astroyctoma and a DIPG appear on an MRI are summarized in the
following table [Table 1].
DIPG Pilocytic Astrocytoma
Diuse Focal
Invasive into surrounding tissue Displaces surrounding structures and
tissue
Diuse brightness on T2 weighting
on MRI; no enhancement on T1
weighted images
Tumor well dened on T1 and T2
weighting MRI
Associated with brainstem swelling Minimal brainstem swelling
Located centrally in pons with
extension to midbrain or brainstem
Located in midbrain and brainstem
without extension
Table 1: MRI Characteristics of DIPG Compared with Pilocytic Astrocytoma
Because the MRI appearance of the two most common types of pediatric
brainstem tumors, DIPGs and pilocytic astrocytomas, are so dierent, they
can be accurately identied the vast majority of the time by MRI alone. In rare
cases where the diagnosis is uncertain based on MRI results, neurosurgeons
can perform biopsies to obtain small amounts of tissue for examination under
a microscope by pathologists (doctors trained to identify the type of tumor
by examining it under a microscope). e biopsy is performed very carefully,
but because the pons contains many important neurologic centers, including
those that control breathing and swallowing, there can be complications of
biopsy, including additional neurologic impairment. For these reasons, biopsy
is generally only performed in cases where the diagnosis is not clear from an
MRI scan. In the future, some clinical trials may include a biopsy to nd out
more information about the tumor prior to starting therapy.
Pathologic grading
Pathologists grade a tumor based on its features. e characteristics that
pathologists examine include cell growth, cell death, invasion of surrounding
normal cells, and the architecture of the tumor itself—this refers to how mature
or immature the cells look, among other factors.
Pathologists grade brainstem tumors on a 1 to 4 scale. e lower numbers
generally indicate less-aggressive tumors, including pilocytic astrocytomas.
e lowest grade consistent with a DIPG is a grade 2 tumor, but many DIPG
tumors will be grade 3 or 4 (the most-aggressive, fastest-growing grades).
Dr. Cohen is Clinical Director
of Pediatric Oncology and
Director of Pediatric Neuro-
oncology at the Sidney Kimmel
Comprehensive Cancer Center
at Johns Hopkins, Baltimore, MD.
Chapter 2: Typical History of DIPG
20
21
Chapter 2: Typical History of DIPG
Clinical Course of DIPG
Once the diagnosis of a DIPG is suspected, anti-inammatory steroids (such
as dexamethasone) are usually started. e steroids can improve symptoms
quickly by decreasing the swelling associated with the tumor. Steroids can cause
side eects including increased moodiness, agitation, weight gain, increased
appetite and high blood pressure and blood sugar. ese last two side eects
can be controlled with medication, if they become severe.
e only treatment that is routinely recommended for the treatment of all
children with a DIPG is x-ray radiation therapy (XRT). XRT can be given
either alone or with chemotherapy and usually takes 4 to 6 weeks to complete.
Side eects during radiation can include mild nausea and fatigue.
Many chemotherapeutic drugs have been tried for DIPG, with studies looking
at the use of chemotherapy before XRT, during XRT, immediately following
XRT, and at the time of tumor progression. e results have been disappointing,
with no drug(s) to date improving survival. While pediatric oncologists continue
to develop new therapies for DIPG, the mainstay of current treatment remains
XRT. ere are ongoing clinical trials for DIPG, which allow new drugs to be
tested in this disease. While there are always risks when enrolling in clinical trials,
they are the best way to get your child the most promising new medications
and to make sure the pediatric oncology community learns all it can about
what therapies work best for DIPG.
Most DIPG tumors in the beginning respond to a combination of radiation
and steroids. e child’s neurologic decits will very often decrease and may
disappear completely. Over the course of weeks to months, the steroids can be
decreased and then stopped in many cases. e child can often return to school,
take special trips, and almost return to normal life. During this time, the child
has regular MRI scans to measure the regression of the tumor and monitor if
the tumor is coming back.
In almost all cases, after about 6 to 12 months, the DIPG tumor starts to grow
again. Sometimes the neurologic symptoms are the same as when the child was
rst diagnosed with DIPG. Sometimes new nerves and systems are aected.
e child will often begin to show neurologic symptoms even if the MRI scan
of the tumor appears largely unchanged.
Once the tumor has started to grow again, no further treatment has been shown
to improve survival. When children start to have neurologic symptoms, they are
often restarted on steroids. is treatment can sometimes improve symptoms for
a short time. However, the tumor will continue to grow, and even if the steroid
doses are increased, the child’s symptoms will continue to worsen. Eventually
the tumor grows until it aects nerve centers that are important for swallowing,
breathing, and controlling heartbeat.
If the tumor is blocking the ow of cerebrospinal uid (CSF), some parents—in
discussion with the doctors—may decide to have a neurosurgeon place a VP-
shunt to help with pressure symptoms. A VP-shunt is a exible plastic tube
that bypasses the blockage in the brainstem and allows the CSF uid to pass
out of the skull. Neurosurgeons place the shunt into the uid cistern in the
brain, and then pass it out of the skull, under the skin, and to the abdomen,
where the CSF is absorbed. is procedure can improve some of the headache
and nausea symptoms of increased intracranial pressure, and it can extend the
life of children with DIPG; it does not, however, change the ultimate outcome.
Only very few children are long-term survivors of DIPG. Because biopsies are
not performed on these children with typical appearing DIPGs, it is unclear
whether or not they actually had DIPG to start with, or in fact had a dierent
tumor or condition that looked like a DIPG on the MRI. ere is no one
treatment that these children received that set them apart from the vast majority
(more than 95 percent) of children who die from DIPG. Pediatric oncologists
are actively looking for new treatments and are trying to learn more about
DIPG. ey hope that by learning more from tumor tissue taken at autopsy
from children who die from DIPG they can help children who develop DIPG
in the future.
Chapter 2: Typical History of DIPG
22
23
Chapter 2: Typical History of DIPG
Parent Perspectives
My son Andrew was diagnosed with DIPG following a couple months of
not feeling well. Initially we thought he had the same virus as his brother.
Perhaps he did, but he did not recover. He was clearly ill—sleeping more
than usual, feeling dizzy and unstable. Someone mentioned to us that it
was funny to watch him go upstairs. We began to notice that his gait was
not right. He looked like he was walking with one foot on the ground and
one foot on a curb, but there was no curb. He struggled to control his
left hand in a piano lesson, and almost fell leaving the studio that day.
When we thought about it, we realized that he had been falling regularly—
either from a standing position or while riding his bike. One night I had
difficulty rousing him from a nap, and had to hold his hand to help him
walk to Children's Church. That same night a nurse in our church noticed
that one side of his face was drooping. By the next morning he was clearly
feeling worse. He could not climb onto my bed, so he lay on the floor in
my bedroom while I called our primary care physician. He vomited, and
we settled him on my bed to rest. When we took him to the doctor that
afternoon, the physician's assistant sent us to the emergency room. All
along they had been thinking it was a virus or a problem with his ears; no
one was thinking it was a brain tumor.
We took our daughter in as a healthy looking beautiful child with only a
bit of drooling, facial numbness, and a bit of balance concern. All of these
symptoms were MINIMAL. If I hadn't insisted on an MRI (the neurologist
didn’t think it necessary but agreed to do it) we wouldn't have found the
tumor when we did. From there it was a whirlwind and you do what you do.
Although it is a blur in some respects, I clearly remember my first thoughts
and words when I was told that my grandson, Miguel had a diffuse intrinsic
pontine glioma. I remember the room clearly. It was a small room in the
PICU area of the hospital. My daughter told me that the doctor said that
if we were lucky Miguel would be with us for two years but the average
survival was around eight months and even then much of that time may
not be good. I remember saying to my daughter, "two days, two months,
two years, whatever it is I'll take it." He was alive at that moment and that
was all that mattered.
In retrospect, there were signs something was wrong but they could mostly
be attributed to normal things (allergies, eating too much too quickly,
tripping over his own growing feet). Over a weekend he began to hold his
head to the side (probably compensating for double vision) so a doctor's
visit was in order. Perhaps there was something wrong with his vision. At
the doctor's office, his pediatrician asked him to lie down and he wouldn't
lay flat. I am sure that he also saw something with the tracking in his eyes.
Miguel's pediatrician decided an MRI was in order and scheduled it for
that same week. Immediately following the MRI Miguel was admitted
to the hospital and placed in the PICU where he stayed for five days
while a plan was put in place. His treatment, we were told, would involve
radiation to try and shrink the tumor. If it responded well, he would have
a "honeymoon period.” His tumor was about 6 mm and the response to the
radiation was nothing short of amazing. All of his symptoms subsided and
he was even able to return to school to finish out the year.
After you’ve swallowed the words “average survival is less than 1 year,”
the next phrase you grasp onto with all of your might is “the honeymoon
period.” Now you don’t know for sure what this is yet but you know
honeymoons are good so, initially you are pretty happy to hear something
that even comes close to positive. As a parent, you find yourself really
looking forward to it as somewhat of a light at the end of a dark and
unknown tunnel.
In the first few days after diagnosis, things tend to be a blur. As we advance
and options change for our kids your road may be slightly different than
mine but for the time being it goes something like this. Your relatively
normal looking child begins to exhibit symptoms of some sort. Eye issues,
balance issues, headache, and nausea, just to name a few. Depending on
how you respond, you eventually find yourself in a pediatric oncologist’s
office where you can feel the air being sucked from your lungs as if you’ve
just had the wind knocked out of you.
Chapter 2: Typical History of DIPG
24
25
Chapter 2: Typical History of DIPG
I decided to take Bizzie to see our pediatrician because she just seemed—
off. She was only two, about to turn three. For the past few months we had
noticed that she wasn’t developing as quickly as we thought she should
be. But, her well visits went fine. She was drooling a little and stumbled
from time to time. Then she started to choke on some foods. Finally, I just
decided to bring her in for a sick visit. My mom was down for a visit, so I
brought her along with me. The pediatrician examined Bizzie. After a few
minutes of looking in ears, mouth, etc. she still did not state any specific
issue. “I’m not worried about her, but…” She talked about sending her in
for a blood test, maybe a CT scan to rule out anything serious. Then she
tested Bizzie’s reflexes. Now she seemed to change her plan. She said she
would be right back—that she wanted to arrange those tests for us.
She left and a minute later someone closed the door to our room. I started
to wonder—that seemed odd. When she came back, she let us know that we
should go right over to our local Children’s emergency room for testing,
that there was a doctor there with whom we should connect.
Our Kayla was diagnosed with DIPG on Aug 23rd. It was her first day
of kindergarten. She had been drooling, she was seeing double, she was
choking sometimes when she ate, and she was getting clumsy. Even a few
months prior I had noticed that she couldn’t keep her right flip flop on when
she walked. So we had an MRI done after our optometrist realized this was
more than a case of misaligned eyes and we were told that Kayla had 6 to
9 months to survive if we did the radiation treatment and 10% chance of
surviving a year with Temodar—a type of chemotherapy. We were in such
disbelief that this could happen to our family but we could clearly see that
she was not well. This was a huge turning point in our lives.
Once this bomb was dropped on us, we instantly went into survival mode.
We came up with hundreds of questions and searched the internet all day
and night trying to understand the disease that we were dealing with.
Courtney was diagnosed with DIPG on April 19th. The only symptoms she
had prior to her diagnosis were fatigue and she had a period of time where
she had dizzy spells about 6 months before her diagnosis. April 14th we
noticed her eye turning in, and took her to the eye doctor on the 16th. He
sent her to the emergency room where they did a CT scan and told us she
had some type of brain tumor but they weren't sure what kind. They sent
us home and told us to come back on Monday the 19th for an MRI. That
is when we found out she had DIPG and that it had spread to her upper
spine. I think we started grieving that day. Not because we didn't hope
that she would make it, but because we knew that the odds were greatly
against her survival. It felt like we walked into that conference room with
a normal life and when we walked out it felt like we had walked into a
whole new world—one that would sometimes feel like a bad dream.
Our son Liam was diagnosed with DIPG in April, just days after his sixth
birthday. The previous month Liam had what we all thought was a nasty
virus. He woke one morning vomiting and continued to do so throughout
the day. At one point I had emailed my husband with concerns that this did
not seem like typical vomiting. It seemed almost more violent than a normal
stomach bug. I remember asking Liam if his neck hurt at all, if anything
else hurt at all, trying to ease the nagging feeling that something wasn’t
right. He ran no fever and the vomiting continued intermittently throughout
the day until evening. The following day the vomiting had stopped but he
suddenly was running a fever. We decided to call his pediatrician who had
us come in. At that time Liam had no other symptoms that were worrisome
to us. His doctor thought that he probably had a case of the flu and did a
rapid test for strep. That came back positive. In hindsight, that was most
likely coincidental and the intense vomiting of the day before was our first
real look at the beast we would come to know so well.
Sam's case is a little on the atypical side—first because he was 19 when
diagnosed, and second because his tumor extended into the cerebellum
from the brainstem. He was fine until January—no hint of anything at all
out of the ordinary. On Jan 11th he woke up with a bad sore throat and
because his band had a show that weekend we went in to our regular doctor
to see if he needed any antibiotics. It wasn't strep but he went ahead and
gave him antibiotics because Sam was prone to sinus infections. Two days
later he woke up with a horrible earache so back we went to the doctor
Chapter 2: Typical History of DIPG
26
who switched his antibiotics. That seemed to do the trick and he felt better
enough by the weekend to perform with his band.
The next week he was really, really tired, taking naps in the afternoon
which was very unlike him. Towards the end of the week he mentioned the
right side of his face was feeling numb. Since it was the same side as the
ear infection we assumed the infection had not cleared up and figured if
it's not better by Monday we'll go back to the doctor. Over the weekend
he mentioned his left leg feeling tingly like it was asleep. That worried
me, but I still thought it would be something minor. So we went to the
doctor on Monday and he said the ear infection was all cleared up and he
had no idea what was causing the facial numbness and leg tingling. He
suggested we see a neurologist and get an MRI so we saw the neurologist
on Wednesday who also noticed an issue with his right eye and agreed
that an MRI was in order. We had that the next day and within half an
hour of being home the doctor called and told us there was a mass on his
brainstem and we needed to see a neurosurgeon.
The next morning Sam was admitted, and we consulted with the
neurosurgeon and the neuroradiologist on Saturday. They explained that
Sam had a pontine glioma that was diffuse in nature with tumor extending
into the cerebellum making it extremely difficult to control. They never
gave us an estimate of time, in fact when we asked they said, "Every case
is different." The radiologist was the most upfront, telling Sam that this
was a very difficult tumor to beat. Sam asked, “Is it possible to beat it?"
and her response after some hesitation was, “Well, it's not impossible."

27
Chapter 3: Pontine Anatomy and Function
Chapter 3
Pontine Anatomy and
Function
Sven Hochheimer, MD
Javad Nazarian, PhD
Suresh N. Magge, MD
Children who are diagnosed with DIPG often experience varying clinical
symptoms. Families are sometimes left wondering why their child exhibits a
particular symptom, while another child may not. Additionally, families may
feel overwhelmed when trying to decipher their child’s MRI, leaving them
unsure how to interpret the ndings on the MRI as they relate to the clinical
signs evident in their child. Understanding pontine anatomy and function
can assist with interpreting MRI reports, as well as explain the variable clinical
symptoms of children diagnosed with DIPG.
General Overview of the Human Nervous System
e human nervous system is divided into the peripheral and central nervous
system (CNS). e peripheral nervous system consists of:
• e somatic nervous system, which is responsible for functions under
conscious control such as body movement and reception of external stimuli;
• The autonomic nervous system, which regulates functions under
subconscious control, such as blood pressure, heart rate, breathing, and
digestion.
e central nervous system is subdivided into the spinal cord and brain, which
includes the cerebrum, cerebellum, and
brainstem. e brainstem consists of the
midbrain, pons, and medulla and serves as
a passageway between the brain and spinal
cord. Above the pons is the hypothalamus,
and to the back sits the 4th ventricle [Fig. 1].
Dr. Hochheimer is a
Neurosurgery Resident at
Walter Reed National Military
Medical Center, Bethesda, MD.

Chapter 3: Pontine Anatomy and Function
28
29
Chapter 3: Pontine Anatomy and Function
Figure 1: Basic anatomy of the Central Nervous System
e pons—which means “bridge” in Latin, is an approximately 3.5 cm. long
“knob-like” structure that occupies the central portion of the brainstem between
the midbrain and the medulla [Fig. 2a]. Any messages descending from the
brain or ascending to it must cross this critical “bridge-like” structure. Anatomy
and function of the pons will be the focus of this chapter.
Figure 2a: Schemata of brainstem cross section
Neurons and Tracts
To best understand anatomy, it is important to gain an understanding of the
terminology of the system being described—in this case the nervous system.
e basic cell of the nervous system is the neuron. Humans have billions of
neurons, yet neurons only make up approximately 10 percent of cells in the
human brain. e remaining 90 percent of cells are support cells called glia.
Neurons
A neuron is composed of dendrites, a cell body, and an axon. Dendrites receive
information for the neuron. e information is then passed through the cell
body and on to the axon. e axon then passes the information along to
dendrites of other neurons. In this way, a neural message gets passed from one
neuron to the next. Axons are covered by myelin, which is produced by glial
cells and serves as an insulation that allows rapid signal transmission.
Collections of neurons that serve a particular function are called nuclei. eir
axons are bundled into collections of thread-like bers called tracts. Tracts that
carry information from the peripheral nervous system up toward the brain are
called ascending tracts, while those that carry signals from the brain to the spinal
Dr. Nazarian is a Molecular
Geneticist and Assistant Professor
in Integrative Systems Biology
and Pediatrics at Children’s
National Medical Center, and The
George Washington University
School of Medicine and Health
Sciences, Washington, DC.
Dr. Magge is a Pediatric
Neurosurgeon and Assistant
Professor of Neurosurgery
and Pediatrics at Children’s
National Medical Center, and
George Washington University
School of Medicine and Health
Sciences, Washington, DC.

Chapter 3: Pontine Anatomy and Function
30
31
Chapter 3: Pontine Anatomy and Function
cord and peripheral nervous system are called descending tracts.
Organization of the pons
e pons consists of a) the basilar pons in the front (ventral portion), and b)
the pontine tegmentum in the back (dorsal portion). e basilar pons and the
pontine tegmentum contain nuclei and tracts [Fig. 2b].e basilar pons contains
a complex combination of tracts (bundles of axons) and nuclei (collections of
cell bodies of neurons). e pontine tegmentum is made up of cranial nerves
which serve the head and neck, associated nuclei, the reticular formation
(neural network involved in functions including cardiovascular control, pain
modulation, sleep and awakening), and tracts (both ascending and descending).
Figure 2b: Cross section of the pons (at the level indicated in Fig. 2a) showing various
tracts and nuclei
Ascending tracts of the pons
e major ascending tracts include the dorsal columns, spinothalamic tracts,
and spinocerebellar tracts, which are described below.
Dorsal columns: e dorsal columns convey information about position sense
(proprioception), vibration, and discriminatory touch. Before reaching the pons,
the bers from these columns cross at the level of the lower medulla to form a
structure called the medial lemniscus, which then traverses the pons. Damage
to the medial lemnisci, at the level of the pons, results in sensory problems on
the opposite side of the body.
Spinothalamic tracts: ese tracts convey sensations of pain, temperature, and
light touch. e tracts cross shortly after entering the spinal cord and do no
t
change sides as they ascend through the pons. Damage to the spinothalamic tracts,
at the level of the pons, results in sensory problems on the opposite side of the body
.
Spinocerebellar tracts: These tracts convey subconscious information
pertaining to proprioception (position sense) to the cerebellum, the part of the
brain concerned primarily with posture, tone, and balance. ese tracts travel to
the cerebellum via structures called cerebellar peduncles. Also, there are several
nuclei within the pons whose axons unite to form one of the cerebellar peduncles
which play a role in the function of the cerebellum. erefore, damage to these
tracts result in problems with posture, tone, and balance.
Descending tracts
e most important descending tracts of the brainstem include the
corticospinal,
corticobulbar, and corticopontine bers, which are described below.
Corticospinal tracts: ese tracts are critical for voluntary movement of the
body. ey originate from the motor areas of the brain and pass through the
basilar pons before crossing at the level of the lower medulla on their way to the
spinal cord. Damage to the brain or corticospinal tract at the level of the pons
results in weakness or paralysis on the opposite side of the body (remember,
these tracts cross to the opposite side in the medulla while on the way to the
spinal cord).
Corticobulbar and corticopontine tracts: Corticobulbar tracts originate in
the brain and control voluntary movement of the muscles of the head and neck.
Corticopontine bers provide a connection between the brain and cerebellum
to coordinate and rene movement. ese tracts also cross, so damage to
corticobulbar bers result in diculty moving the opposite side of the face,
while lesions of the corticopontine bers result in lack of coordination of the
opposite arm and leg.
Other tracts
Two other important tracts, which convey both ascending and descending
information and are prominent in the pontine tegmentum, are the medial
longitudinal fasciculus (MLF) and the central tegmental tract (CTT).
MLF: e MLF is important in coordinating eye, head, and neck movements.
Damage to this tract results in problems associated with double vision, and
diculty with coordination of head and eye movements.

Chapter 3: Pontine Anatomy and Function
32
33
Chapter 3: Pontine Anatomy and Function
CTT: T
he CTT provides an avenue for ascending tracts involved with taste,
as well as provides a path for descending tracts to connect the midbrain to the
cerebellum.
Cranial Nerves and Nuclei of the Pons
As previously mentioned, a number of nuclei (groups of cells that serve a
particular function) reside within the pons. In the basilar pons (front/ventral
portion) reside the pontine nuclei, which serve to connect the brain and
cerebellum. Lesions here result in diculty with coordination of the opposite
arm and leg. Cranial nerves are the nerves that control functions of the head
and neck, and the pontine tegmentum (back/dorsal portion) contains several
of these nuclei.
e lower pons contains cranial nerves (CN) VI and VII [Fig. 3 and 4]. CN
VI, known as the abducens nerve, controls a muscle of the eye known as the
lateral rectus muscle. is muscle allows the eye to move away from midline
toward the temple, a motion known as abduction.
Figure 4: Schemata of the pathways of the cranial nerves
Damage to the nerve results in diculty abducting the eye on the same side
and results in double vision, which is also known as diplopia. e nucleus of
the abducens coordinates the lateral rectus muscle of one eye with the medial
rectus muscle of the other eye, making it possible to move both eyes to the same
side. For this reason, damage to the abducens nucleus results in an inability of
both eyes to look toward the side where the tumor is located, rather than the
double vision seen in abducens nerve lesions.
CN VII, the facial nerve, also resides in the lower pons. Damage to the facial
nerve or its nucleus, results in weakness or paralysis on the same side of the
face. Clinically, this manifests as asymmetric facial expressions and can result in
diculty with eating and speaking. Additional important structures that reside
in the lower pons include the vestibular nuclei, portions of the spinal trigeminal
(the fth cranial nerve, also called CNV) nucleus and tract, and the superior
olivary complex. e vestibular nuclei are involved in balance, so damage here
results in dizziness, vertigo, and postural unsteadiness. e spinal trigeminal
nucleus and tract mediate pain and temperature sensation from the face. Damage
to the spinal CNV nucleus results in sensory disturbances in the face. e
Figure 3: Schemata of the origin within the brain of the cranial nerves
Chapter 3: Pontine Anatomy and Function
34
35
Chapter 3: Pontine Anatomy and Function
superior olivary complex contributes bers to a structure known as the lateral
lemniscus and these structures are involved in hearing. Lesions of the superior
olivary complex or lateral lemniscus result in diminished hearing.
e middle portion of the pontine tegmentum is home to three nuclei (called
the trigeminal nerve) of CNV—the motor, mesencephalic, and principle
sensory nuclei. e motor nucleus controls the muscles that are involved in
chewing, while the mesencephalic nucleus is involved with the position sense
of these muscles. e principal sensory nucleus is primarily involved with
touch sensation from the face. Damage to the motor, mesencephalic, and
principle sensory nuclei result in diculty for the child to coordinate chewing
movements, weakness when chewing, as well as facial numbness. e lateral
lemniscus that forms in the lower pons, and is involved in hearing continues
its ascent through the middle pons. As previously mentioned, damage here
results in diminished hearing.
Other Structures Impacting Function
Reticular formation
e reticular formation is a collection of small neural networks that courses
through the center of the brainstem, including the midbrain, pons and medulla.
It is involved in various functions, including modulation of consciousness, sleep
cycles, pain, posture, tone, and balance. Damage to this critical structure can
cause sleepiness, coma, and death.
Fourth ventricle
e brain has ventricles or cavities that naturally produce cerebrospinal uid
(CSF). is uid circulates throughout the brain acting as a cushion to the
nervous system. In a normally functioning brain, this uid circulates and
provides nourishment to the nervous system, and it is subsequently reabsorbed
into the bloodstream. If the tumor compresses the 4th ventricle, which is
located near the pons, cerebral spinal uid can build up thereby creating
abnormally high pressure within the skull. It is essential that the pressure be
relieved so that the tissues in the central nervous system aren’t damaged and
the blood ow throughout the brain can be restored. Failure to do so can
result in compromised or lost neurological function. Treatment for this uid
buildup—termed hydrocephalus, and also often referred to as “water on the
brain,” is most often achieved through the surgical placement of a shunt or an
endoscopic third ventriculostomy. As is true with the above mentioned clinical
manifestations of DIPG, not all children with DIPG develop hydrocephalus.
Conclusion
An understanding of the anatomy of the pons and the structures it contains
provides the framework for deciphering the various neurological decits that
can be seen in patients aicted with tumors that damage or displace these
structures. A number of problems, including weakness, paralysis, numbness,
incoordination, hydrocephalus, diculties with taste, balance, chewing,
hearing, vision, and disturbances of consciousness, may occur if these important
structures or nearby structures are aected.
Chapter 3: Pontine Anatomy and Function
36
37
Chapter 3: Pontine Anatomy and Function
Parent Perspectives
Every child has a different journey with DIPG. The tumors don't always
grow in the same place, or at the same speed. The brainstem where the tumor
grows controls motor functions. The tragedy of DIPG is that it slowly robs
the patient of motor functions such as walking, arm movements, speech,
sight, eating and breathing while leaving their brain completely intact. That
means that children with DIPG are aware of their decline and continue to
grow and develop cerebrally in every other way.
For Stella, the first thing to disappear was her ability to walk which left
her in August. She has been unable to grasp small objects and do things
like feed herself since mid-September. Her eyesight is compromised, but
she is still able to see some things although we're not sure how much, as
it is difficult to communicate with her. However, Stella's smile remains
intact and the fundamental parts of her personality—fearlessness, humor
and mischievousness are still completely present.
Wednesday May 5th, Ellie stubbed her left toe while swimming with friends.
The next day her tennis coach called because she felt weak and complained
she was not seeing the ball right. It was hot and she said she had not taken in
enough water so we assumed she was a bit dehydrated. Friday she traveled
for the fourth grade field trip for a day of swimming. She had a blast! She
spent the weekend with her good friend playing at the beach.
The following Tuesday on the way to tennis practice she complained her left
leg and arm felt weak. We attributed her left leg to her toe injury but the
arm weakness definitely did not make sense. I called my brother-in-law, a
local physician. He ran through a series of neurological tests and definitely
noted weakness on her left side. We left for the emergency room where we
were met by family and Ellie’s pediatrician. A virus was suspected but a
CAT scan was ordered as a precaution.
Johnny came bounding toward me juice box in hand, when he started to
stumble. His eyes shifted to the left and his head tilted to the left as his gait
fumbled before the fall. He caught himself, but staggered to our meeting
place and finally fell right before he got to me. I helped him up, asked him
if he was alright, made a mental note to call the doctor, and we finished
out a fun afternoon.
That night Johnny had a baseball game and we decided to stop for pizza
before the game. While eating his pizza, Johnny was turned around in his
chair. Always the squirrely kid, Rob and I both told him to turn around
and eat. As he did, he fell out of his chair. We both exchanged concerned
glances since this had happened a few times over the last couple of weeks.
This was not normal. Johnny was our most coordinated child. He was the
one on a skateboard at one, roller skates at one and half, and road a bike
with training wheels before his second birthday. Something wasn’t right.
Again, making a mental note to call the doctor in the morning, we headed
to his ball game.
The next morning I called the doctor’s office to schedule an appointment.
They asked me why I wanted to bring him in and I recalled the incidence
from the day before, stating I thought it might be an inner ear issue. Thirty
minutes later we were at the doctor’s office. The doctor asked me about
the symptoms I noticed. I told him about his eyelids only opening halfway,
giving him a sleepy look. I also recalled his clumsy balance, his appetite
decrease and sleep increase. I had noticed most of these changes within the
last couple of weeks, except his eyelids. They had been slightly drooping
for around a month. Since I had all the kids with me, I was quite distracted
and let the doctor do his assessment without my engagement. I could see
Johnny walking up and down the hallway and the physician’s assistant
shaking her head no, with a concerned look. A few minutes later the doctor
came in and said he was going to schedule an MRI because Johnny had
failed a neurological assessment, and to not leave until he came back with
the time for the MRI. Being unknowledgeable about this, I smiled, agreed
and waited. A few minutes later, he came back in to say we had an MRI
scheduled at 2:30 that day. The doctor also said to not eat anything since
they might want to do sedation. It was starting to sink in. Something was
wrong, very wrong. Before I left, I called my husband to fill him in on all
the details. I also called our parents for support.
As the days went on Liam seemed to recover to a certain degree from the
strep and what we thought was a bug, but never in a way that left us totally
Chapter 3: Pontine Anatomy and Function
38
39
Chapter 3: Pontine Anatomy and Function
comfortable. We began to notice his eyes were at half-mast at times. We
would ask him about it and he promised that he really wasn't doing it
on purpose. These symptoms seemed to come and go. He went to school
every day and overall seemed ok. However as March turned to April those
concerning things we were seeing began to happen more frequently. Liam
came home from school one day with some school pictures. He took them
out to show me and said, "Look Mom, I don't think I know how to smile
anymore." I looked down at his picture and his crooked grin. It was around
this time that things began to change at a rapid pace and Liam's health
changed daily. We become very concerned. His affect began to be flat
and we noticed balance issues. I remember waking up early just to watch
him walking to the bathroom to see how his balance seemed when no one
was watching. He walked the long hallway bumping into the wall and I
remember thinking his walking reminded me of a child with cerebral palsy.
By his birthday weekend our concern grew to alarm. At his birthday party
he did not look well at all. We had watched in a matter of days the brutal
symptoms of his disease come into full view and we voiced our deepest
concerns to family.
Brendan was diagnosed at age 6. Two weeks before diagnosis we noticed
several things. His eyes were cloudy and unfocused which we mistakenly
attributed to conjunctivitis. He had given up napping when he was a
toddler but was very tired and he fell asleep in the middle of the afternoon.
We thought he may be having a growth spurt or was coming down with
something. We noticed he was laughing and talking restlessly in his sleep
where previously he had been a very quiet sleeper. He had difficulty
swallowing and chewed very slowly where he never ate slowly before. His
voice was raspy and he didn't smile anymore. He was experiencing painful
urination and was constipated. He was holding books very close and putting
his face up close to the computer and we and his teacher thought he needed
an eye exam. We finally took him to the doctor when he began falling and
saying he was dizzy. We noticed these symptoms for about 2 weeks before
his falling really alarmed us. Any of the other symptoms alone could be
dismissed as something else but all of it and his falls made it very serious.
We were told his tumor was DIPG. It was in the pons and was typical. They
wouldn't biopsy it because it could cause harm and possibly death, and it
wouldn't change the treatment options available.
He responded well to radiation and was symptom free for 10 months. Upon
progression his eyesight became blurry and he told us he was seeing double
again. He lost dexterity in his right hand so he began to write with his left
until he lost his left side too. He became very clumsy and was falling so we
got him a walker with wheels which he loved to zip around on.
He responded well to re-irradiation and was symptom free for another
6 months. Upon his second progression he experienced the same sorts
of problems with eyes, first his right side then left, and speech, chewing,
swallowing and finally lost neck control and had breathing difficulty. There
were no other options which could give him another “honeymoon.” He
progressed rapidly this time and was gone within 6 weeks of the recurrence
of his first symptom of blurry vision.
Looking back after diagnosis on July 29th I can see things that were red
flags but not nearly as significant as many children have displayed. At some
point near the end of the school year Hope drooled a couple of times and
had food on her face at meal times. I remembered these things that first week
in August but nothing rang any bells then. I remember her brother coming
in the house complaining that she couldn’t even ride a bike straight after
they had been on a ride on their tandem bike. Again nothing tied together
until after her diagnosis.
On July 7th we were watching a little league ball game. Hope sat in a
lawn chair between her brother and me. I looked over and saw that she
had drooled down her chin and onto her blouse. When I asked her if she
realized it she replied, “Mom, how would I feel that? My face has been numb
for 6 weeks.” You can insert a healthy dose of 12 year old attitude to that
statement and then you can imagine my face. As we drove home from the
game several things came to light—like while away for a week at a choir
camp with her best friend, they developed a signal for when Hope had food
on her face. That way she could wipe it off and wouldn’t be embarrassed.
Aimee started complaining of headaches in January, noting that they were
nothing that Motrin couldn’t take care of at first. Then they became more
severe in late February early March. Upon a trip to the doctor, without
tests being done, it was determined she had migraines and was put on meds
for her headaches. Aimee was a cheerleader and had several competitions
Chapter 3: Pontine Anatomy and Function
40
41
Chapter 3: Pontine Anatomy and Function
between May and August and began losing her balance, started getting
hiccups even with just a sip of water. Then she began complaining she
couldn’t breathe. After several more trips to the doctor’s office—again
with no tests done, she was given acid reflux medicine, as well as asthma
medicine. We were told she was dehydrated and to push the fluids. They
also gave her several antibiotics for an inner ear infection. They claimed
that was causing the dizziness and loss of balance. Finally, at the end of
August she began to vomit and needed to sleep often. She told me that the
doctors were crazy, because the only thing wrong with her was that she
had a brain tumor. I of course told her she was crazy, because “kids don’t
get brain tumors.” Then she made a bet with me for $10.00 because she
said she felt it growing.
September 25th, I took Aimee to see a new doctor; he did several
neurological examinations and referred us to a neurologist. The earliest
appointment we could get was in October. Upon leaving the office Aimee
began to vomit profusely, refused to go back in to see the doctor and just
wanted to go home and sleep. She stayed home from school the next day,
and slept and vomited off and on all day. Then on Thursday the 27th, Aimee
woke up with an extreme headache but still wanted to go to school because
it was picture day. Her balance worsened as well. A few days later, we began
to ride our bikes and Aimee was swerving all over the road. I joked that
she was going to get a DUI for her riding. Upon returning to the house I
called the doctor who was not in. His nurse called me a few minutes later
and advised me to get her to the emergency room right away—they are
expecting you.
Gunner was not himself the whole Thanksgiving holiday. He was pretty
lethargic and was complaining of headaches for over a week. He said it was
in the back of his head and moved up and forward to the front. We decorated
for Christmas and he usually is right in there helping, but most of the time
he just laid or slept (which is really unusual for him). He complained of
being hot all the time (which again is not his character). He is usually cold.
He didn’t have a fever at all. He always runs a fever when he is not feeling
well. He also began to just stare off into space—a couple of times while
in the middle of a conversation with me. Gunner started walking and then
sitting down with his head bent down. It made me very uncomfortable to
watch him. He just seemed to have no posture anymore. He started playing
with, and twisting his ears. He also started this mouth swishing thing—it
sounds like when you are washing your mouth out with mouth wash.
I also think he was hard to understand at times. I really had to listen to what
he was saying. Could his speech be getting worse? When I asked Gunner
about his headaches he told me that sometimes he has trouble seeing the
board at school. When I told him that we could get his eyes checked, he got
upset and told me that he was joking. I didn’t think he was joking. He just
didn’t want to go to another doctor appointment. When I tried to calm him
down about his headaches and his eyes, he told me that the TV was split in
half and that when I walked into the room my upper body was not attached
to my legs. He continued to complain of a pain in the back of his head. He
also complained about being dizzy, and tingling in his left hand. I took him
to counseling, a behavior specialist, occupational therapy, an optometrist,
our local doctor, and was given the diagnosis of autism. I felt like we just
kept hitting walls and weren’t getting the help we knew we needed.
Johnny’s tumor is in the pons. It is actually in the back side of the pons
close to the cerebellum. The cerebellum controls motor function. The tumor
is growing and pressing into the cerebellum which is why he is having
difficulty walking and with his balance. The pons controls all his involuntary
functions—his heartbeat, his breathing, swallowing, muscle control and
many other life giving functions. The pons is like grand central station in
the brain. This tumor is taking over those functions and shutting them down.
Julian’s heart rate had slipped into the 30’s in PICU which led to an emergency
midnight ight from Detroit to Memphis. It was one week before Christmas.
That morning, I found myself sitting in the basement of a brand new hospital
waiting to speak with one of St. Jude’s neurosurgeons while Julian was having
an MRI. There was intense debate about whether the dangerously low heart
rate was the result of tumor progression or continued hydrocephalus. Sleep
deprived and practically a shell of myself, our surgeon sat next to me on an
exam table and began to draw a diagram of Julian’s tumor on the sterile white
paper between us.
To my horror, the picture looked like a snake wrapping itself around our beau-
tiful son’s control center as if holding his life for ransom.
Only there was no
bargaining. The site of the tumor controlled his heart rate and respiratory
Chapter 3: Pontine Anatomy and Function
42
func
tion and through the middle ran the critical basilar artery making it too
risky to attempt to surgically debulk. When tears came, I explained that this
was the rst time anyone had shown me the anatomy of this monster. It was
then that the disease earned my respect. It was the most formidable of foes and
it held the upper hand. No drugs had any proven effect. So sly, so cunning, it
reminded me of an evil serpent almost taunting us with its advantage. It was
explained to us that even with intense radiation therapy, we could never kill
every single cell. With even two or three microscopic cells left behind, it would
come back. And when it did, it would return with a vengeance.
Months after we lost Julian, I read how a DIPG researcher described the deadly
glial cell’s shape under a microscope as a preschooler’s illustration of sunshine.
I found it so ironic.

43
Chapter 4: Imaging
Chapter 4
Imaging DIPG
Jonathan Finlay, MD
Girish Dhall, MD
John Grimm, MD
Stefan Bluml, PhD
e understanding of brainstem tumors, including diuse inltrating pontine
glioma (DIPG), has advanced considerably over the last few decades largely as
a result of advances in imaging technology. Nevertheless, the understanding of
brainstem tumors by imaging has always been, and continues to be, limited by
the lack of histopathology (microscopic evaluation of tumor tissue) correlating
to the imaging characteristics. is is primarily related to the risk involved to the
patient through the biopsy of these tumors accompanied by the questionable
direct therapeutic benets for the child.
e development of Magnetic Resonance Imaging (MRI) has signicantly
improved our understanding of these tumors. MRI allows improved
visualization and characterization of brainstem tumors in comparison to
Computed Tomography (CT). MRI provides superior imaging of the posterior
fossa of the brain in comparison to CT, which is limited secondary to artifacts
related to the thick bones of the skull base. Furthermore, in comparison to CT,
MRI has superior soft tissue contrast resolution, which aids in characterization
of these tumors [Fig. 1a, 1b]. e recent development of advanced imaging
techniques including magnetic resonance spectroscopy and magnetic resonance
perfusion imaging will continue to improve our understanding of these tumors.
History of the Imaging of “Brainstem Glioma” and the
Recognition of DIPG
Historically, tumors in the brainstem
were both regarded and treated as a single
entity, namely “brainstem glioma.” In the
early 1990s, as MRI became more widely
available and utilized, several attempts
Dr. Jonathan Finlay is Director
of the Neural Tumors Program at
Children’s Hospital Los Angeles,
and Professor of Neurology and
Neurosurgery at the Keck School
of Medicine, University of Southern
California, Los Angeles, CA.

Chapter 4: Imaging
44
45
Chapter 4: Imaging
were made to characterize brainstem tumors based on MRI criteria. An early
classication system based on MRI was established by Dr. A. James Barkovich of
the University of California, San Francisco, and several of his North American
colleagues. Specically, tumor characterization was based upon:
• the site of primary involvement in the brainstem;
• the longitudinal and axial extent of the tumor within the brainstem;
• the tumor growth pattern (diuse or focal);
• the enlargement of the brainstem;
• and the presence or absence of exophytic growth (i.e. growth extending
outside the brainstem) enhancement, cysts, hydrocephalus, hemorrhage
or necrosis.
Later correlation of such MRI criteria against survival statistics found several
imaging characteristics to be signicant. e particular segment or region
of origin within the brainstem was found to be important. Tumors arising
from the midbrain have the best prognosis. Tumors arising from the medulla
have an intermediate prognosis. Tumors arising from the pons have the worst
prognosis. A diusely inltrative appearance of the tumor was also found to
have a signicantly worse prognosis. A diusely inltrative appearance referred
to tumors that had ill dened margins and involved more than one half of
the brainstem segment of origin, or inltrated the segments of the brainstem
both superior and inferior to the segment of origin (i.e. inltration of both the
midbrain and the medulla if the segment of origin was the pons).
Finally, enlargement of the brainstem by the tumor was found to be negatively
related to survival, with a tumor that enlarges the brainstem having a poorer
survival [Fig. 2a, 2b].
As a result of these ndings, attempts were made to develop new classication
systems that better reected prognosis. Dr. N.J. Fischbein and colleagues of
Germany proposed a classication system in which tumors were divided into 6
groups, each having its own prognostic implications. e six groups included:
1) focal midbrain tumors, 2) diuse midbrain tumors, 3) tectal tumors, 4) focal
pontine tumors, 5) diuse pontine tumors, and 6) cervicomedullary tumors.
It soon became clear that brainstem tumors were a diverse group of tumors
with variable prognoses. Accordingly, management of the tumor needed to be
tailored to the suspected tumor type. Studies in which biopsies were obtained for
larger numbers of patients conrmed this observation. Of note, Dr. Paul Fischer
and colleagues working at Stanford School
of Medicine found that most brainstem
tumors could be divided into two classes:
pilocytic astrocytomas (World Health
Organization grade I tumor) or brillary
Figure 1a: Axial CT demonstrating poor visualization of the tumor, made worse by
artifact from the adjacent thick skull base.
Figure 1b: Axial T2 MRI of the same tumor demonstrates the improved soft tissue
contrast resolution, improving visualization and characterization of the tumor.
Dr. Dhall is an Assistant Professor
of Pediatrics in the Department of
Hematology/Oncology at Children’s
Hospital Los Angeles, CA.

Chapter 4: Imaging
46
47
Chapter 4: Imaging
astrocytomas. Pilocytic astrocytoma had a favorable prognosis (5 year survival
of 95%) and were associated with a location outside of the ventral (anterior)
pons and a dorsal (posterior) exophytic growth. Fibrillary astrocytomas had a
poorer prognosis (5 year survival of 15%) and were associated with a location
in the ventral pons that engulfed the basilar artery [Fig. 3a, 3b].
In addition to these two main tumor types, other tumor types which are less
frequently found within the brainstem include: ganglioglioma, primitive
neuroectodermal tumor (PNET), atypical teratoid rhabdoid tumor (AT/RT),
Figure 2a: Sagittal T2 MRI of a normal brainstem demonstrating the normal anatomy of
the midbrain, pons and medulla.
Figure 2b: Sagittal T2 MRI demonstrating a diuse intrinsic pontine glioma originating
from the pons, diusely inltrating and enlarging the pons.
Figure 3a: Axial T2 MRI of a brillary astrocytoma (diuse intrinsic pontine glioma)
demonstrating a mass arising from the pons, enlarging the pons and engulng the basilar
artery.
Figure 3b: Axial T2 MRI of a long term survivor of a “brainstem glioma” which is likely
not a brillary astrocytoma. is image demonstrates more benign features including a
focal mass with well-dened margins centered in the dorsal (posterior) pons with a dorsal
exophytic growth.

Chapter 4: Imaging
48
49
Chapter 4: Imaging
oligodendroglioma, and lymphoma.
Conventional Imaging
It is now generally accepted that there is a distinct subtype of brainstem tumor
which can be identied on imaging as a diuse intrinsic pontine glioma (DIPG).
ese invariably correspond to brillary astrocytomas on pathology and have
an extremely poor prognosis, the worst of any type of brainstem tumor. ey
demonstrate many of the characteristics that have been outlined above. ose
characteristics include:
• ey generally arise from the pons, more specically the ventral pons.
• ey are diuse tumors, inltrating greater than half the transverse diameter
of the brainstem segment of origin and having indistinct margins.
• ey also tend to expand the segment of the brainstem from which they
arise.
• is expansion coupled with the common location in the ventral pons
results in the tendency to engulf the basilar artery.
• ese masses are generally iso- to hypo-dense (equal to or dark) to normal
adjacent brain on CT, hypointense (dark) to normal adjacent brain on T1
weighted MRI, and hyperintense (bright) to normal adjacent brain on T2
weighted MRI.
• ese imaging characteristics generally reect the increased water content
of these tumors.
• e presence of enhancement after the administration of contrast, cysts
or necrosis, and hemorrhage is variable in these tumors [Fig. 4a, 4b,
4c, 4d]. Necrosis is generally described as areas of relative increased T2
signal and decreased T1 signal within the tumor with peripheral or “ring”
enhancement.
ese typical ndings of DIPG, considered to represent brillary astrocytomas,
are in contrast to focal tumors of the brainstem, which usually represent tumors
of dierent histopathologies.
Several studies focusing on DIPG have
attempted to determine if any conventional
MRI characteristics at the time of tumor
presentation are helpful in predicting
Dr. Grimm is an Assistant Professor
in Pediatric Neuroradiology at
Children’s Hospital Los Angeles, CA.
Figure 4a
Figure 4b

Chapter 4: Imaging
50
51
Chapter 4: Imaging
prognosis. No ndings on conventional MRI at tumor presentation have been
found to be correlated to response to therapy, progression, or overall survival.
Imaging characteristics that have been analyzed include: volume and extent of
tumor, signal intensity on T1 and T2 images, appearance of borders, peritumoral
edema, exophytic components, encasement of the basilar artery, necrosis or cysts,
hemorrhage, gadolinium enhancement, and metastatic disease [Fig. 5a, 5b, 5c].
It is now thought that regardless of the histopathology of the DIPG at the
time of diagnosis, even if the tumor is a low grade WHO II astrocytoma at
diagnosis, most diuse intrinsic pontine gliomas will progress to WHO III
anaplastic astrocytoma or WHO IV glioblastoma multiforme at the time of
Figures 4a, 4b, 4c, 4d: Axial T2 (a), sagittal T2 (b), axial T1 post contrast (c) and sagittal
T1 postcontrast (d) MRI of a diuse intrinsic pontine glioma demonstrating a diusely
inltrating and ill-dened mass of the pons which involves more than one half of the pons
and enlarges the pons, having mass eect on, but not yet engulng the basilar artery. It
is predominantly hyperintense (bright) on T2 and hypointense (dark) on T1 with small
areas of enhancement and possible necrosis.
Figure 4c
Figure 4d
Figure 5a: More images of diuse intrinsic pontine gliomas. Axial T2 MRI demonstrates
a mass originating from and enlarging the pons. Subtle areas of increased signal intensity
(brighter) may represent small areas of necrosis.

Chapter 4: Imaging
52
53
Chapter 4: Imaging
Figure 5b: Axial T2 MRI demonstrating a mass originating from and enlarging the right
side of the pons with ill-dened margins.
Figure 5c: Axial T2 MRI demonstrating a smaller mass originating from the right side of
the pons with ill-dened margins and subtle, if any, enlargement of the pons.
death. Although a small percentage of DIPG will demonstrate areas of necrosis
at presentation, almost all will develop areas of necrosis during the course of
the disease. ere is debate regarding whether the development of necrosis
indicates treatment related changes (i.e. radiation necrosis), or reects the
natural malignant degeneration of low grade tumors (WHO II) to high grade
tumors (WHO III and IV). Although positron emission tomography (PET)
has been used to make this distinction with other brain tumors (see section
below on advanced imaging), there has been diculty in applying this to DIPG.
Nevertheless, many consider the development of new areas of enhancement
or necrosis a grave prognostic indicator that often precedes death [Fig. 6a, 6b,
6c, 6d].
In addition to developing areas of necrosis, most tumors will continue to
inltrate more portions of the brainstem during the course of the disease.
ey preferentially grow in a cranial direction through the midbrain into the
cerebral peduncles and thalamus, and it is suggested that this nding may also
be a poor prognostic indicator.
Figure 6a

Chapter 4: Imaging
54
55
Chapter 4: Imaging
Despite the fact that conventional MRI has not been helpful in the development
of prognostic criteria for DIPG at initial presentation, there continues to be a
small subset of patients that have an unexpectedly long survival. It is uncertain
if the lesions in these patients represent a more benign tumor type other than a
brillary astrocytoma or even lesions other than tumors, such as inammatory
lesions that mimic DIPG on imaging. Furthermore, perhaps these tumors are
less biologically active or are more sensitive to treatment. e advent of new
advanced imaging techniques including magnetic resonance spectroscopy and
magnetic resonance perfusion imaging may soon improve our understanding
of the biology and natural history of
DIPG and lesions that mimic them on
conventional MRI.
Figure 6b
Figure 6c
Figure 6d
Figures 6a, 6b, 6c, 6d: Axial T2 (6a) and axial T1 post contrast (6b) MRI at presentation
and axial T2 (6c) and axial T1 postcontrast (d) MRI one and one half months later
demonstrating the development of necrosis visualized as an area of peripheral enhancement.
Dr. Bluml is an Associate Professor
of Research Radiology at Children’s
Hospital Los Angeles, CA.

Chapter 4: Imaging
56
57
Chapter 4: Imaging
Advanced Imaging
Magnetic resonance spectroscopy
Proton magnetic resonance spectroscopy (MRS) can currently be performed
on most standard MRI machines. e interaction of protons with their
environment helps to identify and measure various substances within the
brain. e most commonly measured substances include:
• choline (Cho, 3.2ppm), related to the synthesis of cell membranes and
increased in states of high membrane turnover (example tumor);
• creatine (Cr, 3.0ppm), a reection of cellular metabolism and generally
used as an internal reference;
• N-acetyl aspartate (NAA, 2.0ppm), located primarily in neurons and con-
sidered a normal neuronal marker, decreased with neuronal destruction;
• lactate (Lac, 1.3ppm), a product of anaerobic metabolism which is in-
creased with abnormal blood ow, abnormal metabolism or necrosis;
• lipids (Lip, 0.9ppm), a constituent of cell walls which is increased as a
product of necrosis and cell membrane destruction.
eoretically, malignant lesions should demonstrate high Cho to Cr ratios and
high Cho to NAA ratios due to the rapid turnover of cell membranes increasing
Cho, and the destruction of normal neurons decreasing NAA. Accordingly,
Smith and colleagues found that MRS may be helpful to distinguish neoplastic
from non-neoplastic lesions in the brainstem, with neoplastic lesions having high
Cho and low NAA levels and non-neoplastic lesions having normal to low Cho
and low NAA levels. Furthermore, MRS may be helpful to dierentiate between
dierent tumor histopathologies when the conventional MRI appearance
is uncertain. MRS may be helpful to suggest other histopathologies such as
pilocytic astrocytoma or PNET, which can often have characteristic proles
on MRS [Fig. 7a, 7b].
Although no metabolic measures on MRS at tumor presentation have been
found to be signicantly associated with survival, several studies have reported
the changes on MRS over time to help to understand tumor biology. Dr.
Laprie and colleagues examined the changes in MRS with radiotherapy. ey
found that Cho to NAA ratios initially decreased within 2 months following
radiotherapy corresponding clinical and conventional imaging responses.
Subsequently, Cho to NAA and Cho to Cr ratios were observed to increase at
the time of relapse. Furthermore, in some patients, changes in MRS preceded
both clinical and MRI deterioration by 2 to 5 months. Similar results were
Figure 7a
Figure 7b
Figures 7a, 7b: Magnetic resonance spectroscopy comparing diuse intrinsic pontine
glioma (a) to pilocytic astrocytoma (b). MRS of a DIPG demonstrating elevation of
myoinositol (mI), choline (Cho), lipid (Lip) and lactate (Lac) with decreased creatine
(Cr) and NAA. MRS of a pilocytic astrocytoma demonstrating a high choline to creatine
level with a characteristically low absolute creatine level and a relatively preserved NAA.

Chapter 4: Imaging
58
59
Chapter 4: Imaging
obtained by Dr. Panigrahy and colleagues who also found that metabolic
changes on MRS preceded clinical deterioration. Serial examination of tumors
demonstrated increasing levels of Cho and Lipids and decreasing levels of NAA,
Cr, and myo-inositol relative to Cho. ese changes likely reect malignant
degeneration and possible transformation from low grade to high grade tumor
[Fig. 8a, 8b].
Magnetic resonance perfusion
Magnetic resonance perfusion imaging (MRP) has recently been studied in
supratentorial gliomas in adults and has been found to be a good predictor
of world health organization (WHO) tumor grade, progression free survival,
progression and death. MRP of tumors is typically performed using dynamic
contrast enhanced perfusion imaging techniques.
MRP techniques rely on the magnetic susceptibility signal loss that intravenous
gadolinium produces on T2* sequences during MRI. By measuring the degree
of T2* signal loss caused by the gadolinium in blood vessels over time, one
can determine the relative volume of blood, or relative cerebral blood volume
(rCBV), within a tumor. Similarly, the relative cerebral blood ow (rCBF)
and the mean transit time (MTT) can also be measured. ese measurements
Figure 8a
are thought to be surrogate markers for blood vessel proliferation within
malignant brain tumors—an important feature in the grading of gliomas on
histopathology. Accordingly, several studies have shown that gliomas with low
rCBV, and theoretically lower tumor blood vessel proliferation, have longer time
to progression and gliomas with high rCBV, and theoretically higher tumor
blood vessel proliferation, have shorter time to progression. is was found to be
true regardless of WHO grade on biopsy. Some studies have even suggested that
measurements of rCBV were more predictive of prognosis than histopathology
and WHO grade on biopsy. It has been proposed that this nding is most likely
the result of either sampling error on biopsy (as tumors are heterogeneous and
the portion of the tumor with the highest WHO grade may be missed on biopsy)
or variability in consistency of diagnosis among pathologists.
To date, little research has been performed utilizing MRP in the analysis of
DIPG. is may be a promising avenue to help stratify patients based on the
aggressiveness of their tumors and their prognosis [Fig. 9].
Figures 8a (previous page), 8b (above): Magnetic resonance spectroscopy demonstrating
signs of progression with interval increase in choline (Cho) and the choline to creatine
(Cr) level over a period of 5 months from 8a to 8b.

Chapter 4: Imaging
60
61
Chapter 4: Imaging
Positron emission tomography
Positron Emission Tomography in combination with CT (PET/CT) is a tool
that helps radiologists understand the metabolic activity of tumors. PET/CT
measures the accumulation of tracers in cells that are incorporated into the cells
during metabolism. e most commonly used tracer in PET/CT is 2[18F]
uoro-2-deoxy-D-glucose (FDG). FDG is a measure of glucose metabolism,
and most tumors have increased glucose metabolism in comparison to normal
tissues. A less frequently used and less widely available tracer in brain tumor
imaging is 11C-L-methionine (MET), which is involved in amino acid
transport, also increased in tumors. MET may be more sensitive for low grade
tumors, in comparison to FDG, due to the lower background activity of MET
in normal brain.
Several studies focusing on brain tumors have shown that PET/CT can help
to separate less aggressive tumors from more aggressive tumors, tumor from
radiation necrosis, and tumor from scar tissue. Fewer studies have focused
exclusively on brainstem tumors in children. ese studies have suggested that
PET/CT may also be helpful in the evaluation of DIPG to dierentiate low
grade tumors from high grade tumors, with FDG uptake being increased in high
grade tumors. Dr. Kwon and colleagues found that only WHO IV glioblastoma
multiforme tumors were FDG hypermetabolic (“hot”) [Fig. 10a, 10b, 10c].
Furthermore, Dr. Pirotte and colleagues noticed that patients with the highest
FDG uptake had shorter survival times. PET/CT has also been found to
be useful in planning biopsies. In comparison to utilizing contrast MRI for
planning biopsies, PET/CT has a higher diagnostic yield, requiring a fewer
number of biopsies. Furthermore, PET/CT guidance yielded an equivalent
or higher tumor grade in comparison to MRI guidance. As mentioned
previously in the section on perfusion imaging, DIPG are heterogeneous in
their histopathology, and PET/CT can help to guide a biopsy to the areas of a
tumor with the highest grade. is will help to prevent the under-grading of a
tumor with a conventionally guided biopsy. ere have only been a few small
studies of PET/CT in brainstem tumors and further work is needed to develop
this promising technology.
Figure 9: Magnetic resonance perfusion imaging of a diuse intrinsic pontine glioma
measuring relative cerebral blood volume. e graph plots the signal intensity of the
tumor over time. e signal intensity decreases when the contrast bolus enters the blood
vessels within the tumor.
Figure 10a: Axial T2 MRI (a) demonstrating a diusely inltrating mass involving the
left pons and left cerebellar hemisphere.

Chapter 4: Imaging
62
63
Chapter 4: Imaging
Figures 10b, 10c: Corresponding axial CT (b) and axial FDG PET (c) demonstrate a
hypometabolic (cold) lesion.
Figure 10b
Figure 10c
Summary
Advances in imaging techniques over the last several decades have improved
our understanding of brainstem tumors. It is now generally accepted that there
is a specic subtype of brainstem tumor that can be identied on imaging as
a diuse intrinsic pontine glioma (DIPG). is tumor invariably represents a
brillary astrocytoma on histopathology and has an extremely poor prognosis—
the worst of any type of brainstem tumor. On imaging this tumor is typically:
• T2 hyperintense (bright);
• arises from the pons;
• is diusely inltrating with ill-dened margins;
• involves more than one half of the pons;
• expands the pons often engulng the basilar artery.
Despite advances in imaging technology, we have yet to nd imaging features
that can help to predict prognosis. New imaging techniques, including magnetic
resonance spectroscopy (MRS), perfusion imaging and positron emission
tomography (PET), may someday help us to better understand the biology of
this tumor, and in turn improve the treatment of this tumor.
Chapter 4: Imaging
64
65
Chapter 4: Imaging
the time to try and show me comparative scans—then and now from the same
angle. I saw some stuff I didn’t like but I also saw the reduction in size in spots
and some clean edges of the brainstem where before I saw that diffuse haze. It
was very good to refuel my hope.
When her tumor started to progress a month later she was rescanned. I knew it
was back. This time they scanned her spine and it had spread there too. I didn’t
need to see any more scans at that point in time.
Tumor measurements were very important to us following the rst few scans.
It was six months after diagnosis when we began to realize that measuring a
diffuse tumor was not an exact science. The cancer cells are sprinkled among
healthy brain cells. So how do you know where to start measuring? It was about
this time that we understood the importance of having the same radiologist
read and compare scans. One radiologist’s measurements may differ substan-
tially from another’s. We also realized by this time that DIPG scans need to be
read by someone who has a good amount of experience with DIPG. We got a
copy of every scan and sent it to Andrew’s neuro-oncologist for her opinion.
I feel that the MRIs that Noelia had done were sufcient to get an accurate
diagnosis. I personally did not try to read them though. I listened to our doc-
tors explain them to us (usually the day after the scan was done) and I have re-
viewed the written reports which did not give me any more information I would
understand. Our doctors did a great job explaining each scan. They took the
time to answer all of our questions and even explain the scans to other family
members that had questions in Spanish. My rst impressions were mind numb-
ing though, because the doctors we spoke to were very direct in the prognosis
of the tumor. They did not try to sugar coat it. I understand that there is no cure.
I understand it’s inoperable. I understand that there is no reason why. Noelia’s
tumor was located in the middle of the pons and we could see it very clearly
in the scans. We also saw the change in the tumor after radiation. It reduced
in size about 50 percent, but obviously it grew back. We did not have any more
scans done after the symptoms came back. We knew what was happening.
Parent Perspectives
I think that imaging is important in this diagnosis because it can be very risky
to do a biopsy. However I highly recommend sending scans to a few other doc-
tors that deal with this tumor regularly for their opinions. We did try to read
and understand the scans and it was difcult and overwhelming often. I would
try to print off comparable images from different scans. A few times our doc-
tors did pull up our scans right at the hospital to talk to us about them directly.
Our doctors were very kind and spoke to us about our child’s prognosis (when
it was good and bad) while reviewing the scans with us.
When Tatumn was diagnosed we asked for copies of the MRI scans so we could
send them to other centers for second opinions. I felt a little strange about di-
agnosing her tumor with just an MRI. My personal background taught me the
only way to really be sure of cancer is a surgical biopsy. But all of the doctors
were in agreement that this was a DIPG and this was how they did it now. To
be sure I had them sent to other hospitals and received the same response.
After being home from the hospital a week or so we decided to open one of
the discs on our own computer because we were still trying to wrap our head
around what was going on in her sweet little head. So we downloaded a pro-
gram to open the scans on our computer. A lot of the pictures were hard for
me to decipher and I was trying so hard to remember what the doctors had
explained to me on that overwhelming night.
I found what I call a prole shot. It showed her tumor from the side of her
head. I saw that hazy mass and was overtaken by nausea. That picture was so
hurtfully powerful and so ugly at the same time. I knew I couldn’t look at any
more of the pictures and neither could my husband. We never opened them
again. When she had her rst scan after radiation, the doctors were excited
to tell us that there was about a 25% reduction. Since there are so many pic-
tures and angles I couldn’t sort through everything I saw. So I asked to see an
original diagnosis image and a matching after radiation image from the same
perspective. I wanted to see a difference. I don’t know why I had to ask that
and it wasn’t just shown to me that way. Nonetheless our neurosurgeon took
Chapter 4: Imaging
66
67
Chapter 4: Imaging
Fear was written all over Johnny’s body, as he faced the unknown of an MRI.
An MRI requires the patient to hold very still for at least fteen minutes. The
technician offered two options. Johnny could have sedation but I would not lay
on the table with him or he could go without the sedation and I would lie on the
table during the MRI. Johnny chose to have me on the table and gave up the
sleep medicine. The tech explained everything that was going to happen and
about how long it would take.
As he lay down on the table, they put a brace around his head with a mirror
on it. Instead of moving his head to see me, he could look in the tilted mirror.
It is a small tube that you slide into and when they turn on the machine the
magnets bounce around making a horrendous noise. My husband and I were
given ear plugs. Johnny had on headphones and was able to listen to the radio.
As I lay on the bed, I watched as my seven year old son showed more tenacity
than most adults. He lay there with his eyes closed. Periodically he would open
them, and look at me. I would smile and he would smile. I would wink and he
would smile. I would close my eyes and he would return to closing his eyes.
At one point he even fell asleep and I watched as his breathing evened and his
chest rose in rhythm. The noise was deafening even with ear plugs in. I didn’t
nd out until later how terried he was and how much comfort I was to him.
The magnets slowed in their bounce, the technician came in and told us he did
a great job and we headed toward home.
In the morning, we had an appointment to meet with the doctor. As we entered
the doctor’s ofce we were taken to a room with large windows. The room had
a wooden porch bench, a computer, a stool and a patient table. We sat down
on the wooden bench with the doctor sitting on the stool next to the computer,
and the physician’s assistant standing on the other side of the monitor. The
MRI scan was displayed on the computer. The doctor explained that there was
a mass around four centimeters in the middle of the brain called the pons. He
gave us a photocopy of a brain and circled the pons. He went on to explain,
“This area of the brain controls all the involuntary functions of the body—ev-
erything a body does without thinking.”
We nodded with partial understanding and amazed disbelief. He turned on the
monitor to show us the scan. He showed us exactly where the mass was, outlin-
ing the perimeter. The scan was very clear showing to us that Johnny had done
a great job holding still. There were multiple frames and angles of the brain
and the mass. The doctor asked if we had any questions. We were speechless.
It was an unbelievable moment to nd out that our son had something in his
brain and we would drop life to do anything to x the problem so he could be
healthy again.
We learned that Johnny had a cancer called DIPG. The tumor is inoperable
and the cancer is incurable. At the sound of those words, I buried my face in
my hands. In the background I heard details about radiation and experimental
chemotherapy. The doctor recommended not taking a biopsy or attempting to
remove the tumor due to its location, but if we did want to pursue that option
there was a hospital in France that was taking biopsy tissue. We both nodded
in agreement that this was not a course of action we were interested in pursu-
ing. After answering questions, he briey discussed the protocols available
and we began an eight week treatment plan.
Upon discharge, we were given a CD copy of all of his scans. We went back
every eight weeks for a follow up MRI to monitor tumor changes.
We met with Liam’s neurosurgeon who showed us the CT scan done the day
before. This was our rst time seeing the image. I knew almost instinctually
that this could very well be our worst case scenario playing out. The doctor
pointed out the lesion and then the area around it. Liam’s doctor said that area
was what we needed to understand better and would with an MRI. His words
and direct eye contact struck me. I turned back to the image and couldn’t take
my eyes off that area.
Later that same day, Liam had his MRI. We were given the news the next morn-
ing. We were taken to a small room lled with at least four doctors, a nurse and
a social worker. The MRI image was brought up on a screen. It was clear it
was in his brainstem. His neurosurgeon told us it was a brainstem glioma that
originated in the pons of Liam’s brain. It was inoperable. The rst words from
my mouth were: “How long do we have with him?” Our doctor answered,
“Possibly a year but most likely less.” As strange as it may sound I felt relief.
Based on how quickly Liam’s health had declined and how sick he was when
we brought him to the hospital I had thought surely he had, at the most, only
months. I was trying desperately to squash the terror that we may not be able
to even bring him home from this place. A year seemed, in this strange and ter-
rifying new world, like a blessing. It was our rst lesson in the gift of perspec-
tive that this journey gave to us.
Chapter 4: Imaging
68
69
Chapter 4: Imaging
Around November, I began to notice very small changes—changes even Liam’s
Dad did not sometimes see without me pointing them out. Soon though, he
began to notice them as well. Intermittent vomiting returned. Strangely this
came with no nausea or warning and once nished did not bother Liam at
all. He took to calling it his “party trick.” In December and early January
we noticed balance issues here and there. He was scanned after the holidays
but his scan appeared relatively stable. We did not believe this was the case
based on what we were seeing at home and we were blessed to have doctors
that listened when we voiced our concern. We were told, “If mom or dad tells
us that something is not right, we believe them. That’s all we need to hear. We
will always treat Liam not his scan.” How blessed were we! It was decided
to rescan Liam in 4 weeks vs. the two months that was previously the norm.
We had learned that sometimes a scan doesn’t always reect how a child is
presenting clinically, especially with DIPG. It’s that old sand analogy again.
Hard to sometimes visualize the subtle changes that are occurring in the brain
on a scan but that are clear as day when spending time with the child himself
and especially when viewed under the VERY, and by this point, practiced and
careful eye of his parents! In four weeks, Liam’s scan matched what we were
seeing in our every day. Nine months from diagnosis the “honeymoon period”
was over and tumor progression had settled in.
Initially our son was given a CT scan of the brain ordered by his local pedia-
trician. This image only showed the area of necrosis within his tumor and an
area of abnormality around that. However, the area of concern around the
“lesion” could not be denitively diagnosed without an MRI which was done
two days later.
In subsequent images taken, the MRI in general was a good marker for prog-
ress. However, later when his symptoms slowly started to re-appear his MRI
image did not reect what we were seeing symptomatically. Thankfully his
doctors recognized that it can take the scan image some time before it reects
what a parent may be seeing clinically.
We were very fortunate that the same person sat with us every time to review
his scans. This is not always the case and can be problematic at times. It’s im-
portant to establish continuity of care as much as possible. It’s also advisable
to have someone (maybe not too many doctors though) review your child’s
scan who is very experienced in reading scans of DIPG kids.
We were extremely fortunate to have our son’s neurosurgeon read and review
every one of his scans with us personally while he was recovering from anes-
thesia. At times the radiologist also called us in if there were any areas she had
a particular concern over so that we might be able to discuss them then and
not wait. Because this time was taken, we always walked away from each scan
with a peace and understanding about what the results said. This practice is
not the most common but if possible can do so much to aide in the continuity of
care for families. It was a blessing in the midst of so much uncertainty.
We took Gabby to see her pediatrician after she was having problems with
her balance and her speech seemed off. The pediatrician didn’t seem too con-
cerned with her reex test as well as her balance, but she wanted to keep get-
ting her tested to ease our minds. We were scheduled for an MRI a week later
and in that time we went for blood tests and an eye exam. Over the following
weekend Gabby’s balance got so bad that she was holding on to furniture to
walk. We decided not to wait the extra day for her MRI and took her to the
emergency room. She had a CT scan in the emergency room that day and we
were told there was some uid buildup but they were not really impressed by
it. She was going to be admitted and they would schedule her MRI for the next
day. When we went for the MRI they decided since she would be sedated they
would do a lumbar puncture as well. After the MRI while she was in recovery
from the sedation they had decided against the lumbar puncture and were
sending her back to her room.
That’s when we rst knew something was wrong. The same doctors we saw in
the emergency room came to deliver the news to us that she had a tumor and
it was not good. They were assembling a team and they would be back in an
hour to discuss it with us. Left in total shock and disbelief, my husband and I
waited more like 3 hours to hear from the doctors again. They brought us into
a room and showed us her slides from the MRI. They described the tumor as
being in the pons of her brainstem and diffuse so they were calling it DIPG,
but it was also in several other areas of her brain that made it atypical for this
disease. They recommended an additional MRI with contrast, and a biopsy of
her cerebellum to be sure we knew what we were dealing with. The additional
MRI did not show anything else.
Chapter 4: Imaging
70
I sat in the balcony for the nal performance of a play that Hope was in that
summer. I was surprised to see her stumble over a simple dance step but no
one else noticed and she didn’t miss a beat so I let it go. On Sunday night I
was not connecting this to the other symptoms. I would begin to do so by Tues-
day. On Monday the neurologist also thought I was crazy. He reiterated our
family doctor’s diagnosis saying that she had Bell’s palsy, that it would take
some time, but if I was worried he’d order an MRI to make me feel better. I
remember very clearly seeing him backing out of the room smiling, somewhat
like I was a crazy over-protective mother, saying he was sure this was nothing.
As “luck” would have it there was a cancelation at the MRI suite at our local
hospital on Tuesday. We took it thinking it would be great to get this over and
done with so we could move on with our summer. On Tuesday, the look on the
MRI technician’s face—who practically held my hand as he walked us to the
door—spoke volumes. He sent us directly back to the neurologist’s ofce who
gave us the news that there was a “mass” on the brain.
When Sam was rst diagnosed and we were in the hospital the neurosurgeon
made a point of showing Sam and my husband the MRI and the tumor and
where he was going to remove what he could. I was not there at the time so
missed that. Later when we had MRIs done after the radiation and when he
was on chemo the neuro-oncologist always showed us the MRIs but he went
so fast from view to view. I understood what he was showing us at the time
but could never nd those views once I got home with the CD. We have all the
MRIs on a CD along with the radiologist’s reports but there are hundreds of
views on the CD for each MRI done and I can’t gure out which ones are the
ones we looked at in the hospital. It was frustrating at the time and still is a
source of frustration that I can’t see the images of his tumor on the CD.

71
Chapter 5: Clinical Trials for DIPG
Chapter 5
Clinical Trials for DIPG
Adam Cohen, MS, MD
Howard Colman, MD, PhD
Many researchers are working hard to develop new treatments for DIPG. To
date, no therapies are available that provide a lasting cure for this disease. As
new treatments become more readily available, parents will be given additional
opportunities to enroll their children in clinical trials. is chapter is focused on
clinical trial design. Parents who are familiar with study design terminology and
who have a basic understanding of the purpose of clinical trials are better equipped
to make informed treatment decisions. Every child is unique, and treatment
decisions are also unique to individual children and families.
What is a Clinical Trial?
A clinical trial is a way to test possible new treatments. Clinical trials are done in
cancer to nd out:
• whether a treatment is safe;
• whether a treatment has the eects on cancer cells that scientists think it will;
• whether it is eective at shrinking tumors, delaying growth of tumors, or making
people live longer.
A clinical trial is a scientic experiment that must follow certain rules to ensure patient
safety and that the results are true and not due to chance or bias. A clinical trial may
test several dierent types of experimental treatments including:
• A brand new drug (usually not already FDA approved for other cancers or uses).
• A new combination of drugs (that could include new or existing drugs).
• A new technology.
• An old drug that is being used in a new
way or on a new population of patients.
Dr. Cohen is an Assistant
Professor in the Division of
Oncology, and Department of
Internal Medicine at the Huntsman
Cancer Institute, Salt Lake City, UT.

Chapter 5: Clinical Trials for DIPG
72
73
Chapter 5: Clinical Trials for DIPG
ere are many types of clinical trials. When participating in a clinical trial, it is
important to know:
1. What kind of trial it is.
2. What the purpose of the trial is.
3. What is new and experimental about the trial.
Clinical trials are an essential part of medical research. Without clinical trials, medical
knowledge cannot grow and new treatments that may save or improve the lives of
future cancer patients would not be identied.
What a Clinical Trial Is and Is Not
Clinical trials must have specic characteristics that qualify them as well-
designed scientic experiments. Not every study is a clinical trial. In this
section, we give examples of studies that are not clinical trials and give some of
the qualities of a good clinical trial.
Clinical trials must study a predetermined group of people, given a predetermined
treatment and use statistics to analyze results. “Case reports” or “case series”
are reports by physicians giving the results of a specic treatment observed in
just a few patients. While potentially useful to other physicians, case reports
of one or two patients are not true clinical trials because not enough patients
are treated with a specic therapy to know if the results are due to luck, to the
treatment given, or to some unknown factor. Because everyone likes a happy
ending, examples of patients who seemed to respond to a treatment are much
more likely to be published than examples of patients who did not respond to
a treatment.
Retrospective series are also not true clinical trials, but generally provide more
data and reliability than case reports. Retrospective studies look backward in
time to study a group of patients with the same disease. Retrospective series
are not clinical trials because the group of people and the treatment were not
dened before the treatment was given. erefore, the results may be biased
by the choices both doctors and patients
make. For example, healthy people tend
to be treated aggressively, and healthy
people tend to live longer; but that does
not mean that more aggressive treatments
make people live longer.
A well-designed clinical trial needs to include:
1. A clear research question.
2. A hypothesis.
3. A specically dened population.
4. A description of the treatment so that everyone in the trial gets treated the
same way.
5. A follow-up plan that describes how often the eects of the treatment will
be reassessed.
6. A statistical plan that describes how to determine at the end of trial, the
answer to the initial question.
A clear research question describes the purpose of the clinical trial. Examples
of clear questions are:
1. How often do people taking this drug experience severe side eects?
2. How often do tumors shrink when people take this drug?
3. Which drug makes people with a brain tumor live longer?
A clear question prevents misinterpreting unexpected results that could be due
to chance.
e hypothesis is the researcher’s educated guess at the answer to the question.
e hypothesis spells out how safe or eective a treatment should be for the trial to
be considered a success. e hypothesis also makes it clear how to know if the trial
does not succeed, so that unsafe or ineective treatments are not studied again.
Clinical trials are conducted in specic, well-dened populations. e
population may be limited by specic diagnoses (cancers), ages, symptom
levels, overall health, or other factors. ese limitations are called the inclusion
and exclusion criteria. For example, a clinical trial of brainstem gliomas may
allow adults and children, just children under 18, or just children under 12,
etc. Most, but not all, clinical trials require participants to have normal liver
and kidney function and exclude patients with other major medical problems
or multiple cancer types. Limiting the population for clinical trials helps to
ensure the scientic validity of the trial. However, clinical trial results may not
apply to people outside of these limits. For instance, results from a clinical
trial allowing adults with brainstem glioma may not apply to children with
diuse intrinsic pontine glioma if they were excluded from that trial.
Dr. Colman is an Associate
Professor and Director of
Medical Neuro-Oncology in
the Department of Neurosurgery,
at the Huntsman Cancer
Institute, University of Utah,
Salt Lake City, UT.
Chapter 5: Clinical Trials for DIPG
74
75
Chapter 5: Clinical Trials for DIPG
A clinical trial must have a treatment plan, follow-up plan, and statistical
plan set out before starting the trial. is ensures that everyone in the trial
is treated the same way and that the doctors running the trial cannot change
things to make the trial results appear better than they actually are. It also
potentially limits the treating doctor because tests or treatments may need
to be done at specic times that are less exible than they would be with
treatments outside of a clinical trial.
Clinical Trial Phases
Clinical trial phases correspond to the dierent stages of testing that a drug goes
through before approval. For people with brain tumors, the clinical trial phases
are phase 0, phase 1, phase 2, or phase 3. e phases are sometimes referred to
using the Roman numerals 0, I, II, III.
Phase 0 studies
Phase 0 clinical trials are usually done very early in the testing of a new drug.
Phase 0 clinical trials test whether a drug gets into the brain or brain tumor
cells and whether it “hits” the expected target. For example, if a new drug is
developed to inhibit the growth protein EGFR, a phase 0 study can look at
the activity of the EGFR protein in the brain tumor before and after treatment
with the new drug. Phase 0 studies usually require surgery to obtain a sample
of the tumor after treatment with the drug; however, depending on the specic
drug and the expected target, testing of drug eects can sometimes be done
on blood samples or other tissues. A common way of doing phase 0 studies is
to have someone take a drug for a week before a planned surgery and then use
some of the tumor removed during surgery to determine the eects of the drug
on the tissue. One important aspect for patients to understand about Phase 0
studies is that they generally are not expected to benet the individual entering
the study. ese studies are often done very early in the development of a drug
before the optimal dose is determined, and treatment is often continued only
for a short period of time. ese studies do, however, potentially allow for a
better understanding of how a drug works; this, in turn, aids in the planning
of further trials. Phase 0 studies are generally not applicable for patients with
inoperable tumors, such as diuse intrinsic pontine gliomas, which some believe
cannot be biopsied safely.
Phase 1 studies
Phase 1 clinical trials are done early in the testing of a new drug or new treatment
combination to nd what doses of a drug are safe to give. Phase 1 clinical trials
often also test how long the drug stays in the body and how the body processes
and gets rid of the drug. ere are sometimes a number of extra tests required
in phase 1 studies, such as blood tests, urine tests, EKGs of the heart, etc., that
test for toxicity to dierent organs and to see how long the drug is staying in
the body and how it is getting out of the body.
For traditional chemotherapy drugs, phase 1 studies often have the goal of
dening what is called the Maximum Tolerated Dose, or MTD. e maximum
tolerated dose is the dose just below where a high proportion of people have
unacceptable side eects from a drug. e maximum tolerated dose is the highest
dose that should be used for that drug because higher doses are generally too
toxic. Sometimes, particularly for modern drugs that target specic molecules
in cancer cells, phase 1 studies do not go all the way to the maximum tolerated
dose, but stop at a dose where the drug is expected to completely inhibit the
molecule it is targeting. is is called the Biologically Eective Dose.
People are enrolled in phase 1 studies as part of a group, which typically is called
a cohort. Cohorts usually consist of three people but can vary in size from 1
person to many people. Each cohort is given a dierent dose of the drug being
tested. e rst cohort gets a very low dose. If the side eects are tolerable,
the next cohort gets a higher dose. is continues until too many people in
a cohort get intolerable side eects. ese intolerable side eects are called
Dose Limiting Toxicities (DLT). e highest dose whose cohort did not get
intolerable side eects is the Maximum Tolerated Dose (MTD).
Phase 1 trials are a very important step in developing new cancer drugs, and
everyone in a phase 1 trial is helping patients in the future. Traditionally Phase 1
studies have not focused on how well a drug works—just the safety and dosage.
In most phase 1 studies, once you are in a cohort, the dose of the drug will
stay the same for you. Other people who are in other cohorts may get a higher
dose of the drug. erefore, people in the earlier cohorts of a phase 1 study
may get a dose of the drug that is below the dose that is thought to be needed
to shrink the tumor. For this reason, as well as the fact that most drugs tested
in Phase 1 will not go on to demonstrate signicant ecacy in future studies,
the expectation is that most people in a phase 1 trial are not likely to personally
benet from the treatment being tested. erefore, phase 1 trials are often most
appropriate after patients have failed standard treatments with proven benet
or when there are no proven treatment options available.
However, as drug companies have become more focused on drugs targeting
specic proteins and molecular pathways in tumors, the role of Phase 1
Chapter 5: Clinical Trials for DIPG
76
77
Chapter 5: Clinical Trials for DIPG
testing has started to change. In the case of these drugs, the specic targets
are sometimes known before the study. us, it is possible in these specic
situations to hypothesize that patients whose tumors have that specic molecular
target may be more likely to respond to that specic drug. So, in addition to
dening the Maximum Tolerated Dose, some more recent Phase 1 studies also
include initial measurements of tumor response or drug eects on tumors.
is approach has been used successfully by scientists to demonstrate potential
ecacy of promising drugs early in their development, which has accelerated
the subsequent Phase 2 and 3 studies and FDA approval. It is possible that in
the future, patients participating in Phase I trials of targeted drugs may have a
higher chance of getting benet from the treatment in the situation in which
the patient and the physicians know that the patient’s tumor has that molecular
target.
Phase 2 studies
Once the safe dose of a drug is known, phase 2 clinical trials are used to see if the
drug is eective against a certain kind of tumor. Phase 2 trials may have many
dierent designs. In some Phase 2 studies, called single arm studies, everyone
gets the same drug and dose. In multi-arm or randomized studies, there are
several dierent treatments being tested or compared. Patients may get a specic
treatment based on particular criteria of the study, or patients may be randomly
assigned to treatment arms. Randomly assigned, also called randomization,
means that neither you nor your doctor can choose what treatment you get.
Such randomization is necessary because otherwise conscious or subconscious
biases can inuence the results of a trial. For example, if doctors gave everyone
with small tumors the new drug and everyone with big tumors the old drug,
then the new drug might look better even though it might not be better than
the old drug.
e goal of a phase 2 study is to see if a new drug or combination of drugs
has some benecial eect on the tumor or other preferred patient outcomes.
However, even though some Phase 2 trials include more than 100 patients,
they are generally not large enough to conclusively prove whether people live
longer when they take the new drug or to prove absolutely that a new drug
helps. Instead, phase 2 trials often look at endpoints other than how long people
live that can indicate whether a drug is helpful. ese alternate endpoints can
include:
• Response rate: the percent of tumors that shrink a certain amount (usually
25%) with a treatment.
• Disease control rate: the percent of tumors that shrink or stay the same size
with a treatment. Because of random variation between scans, tumors must
grow by at least 20% to 25% to not be considered the same size. us, if a
tumor grows 10% or shrinks 10% it is considered stable, i.e., the same size.
• Progression Free Survival (PFS): the amount of time from the start of
treatment until someone either dies or the tumor progresses, which means
it grows more than a set amount. PFS is often described as a median, the
amount of time until half of people die or have tumor progression. For
example, if the median PFS is 6 months, then by 6 months after the start
of treatment, half of the patients have either died or had their tumor grow
and half of the patients are alive with tumors that are either the same size
or smaller.
• PFS3 or PFS6: the progression-free survival at 3 months or 6 months
respectively, after the start of treatment. is is the percent of people who
are alive with tumors that are the same size or smaller at the specied time
point.
While it seems intuitive that treatments that shrink tumors or that delay tumor
progression will make people live longer, there are occasional cases where this
has turned out not to be true. For example, sometimes tumors can grow back
faster after a drug stops working or a tumor may appear to shrink on an MRI
but the cells are continuing to grow and invade new parts of the brain. Because
the study is testing how eective a drug is, patients participating in a phase 2
trial have a higher chance of personally beneting from taking the drug than in
earlier Phase studies. However, trial participants may not benet if the treatment
turns out to be ineective or if the treatment only works at certain doses or
in certain people. Often as part of a phase 2 trial, tests are done on tumor
specimens or on blood samples to try to identify which people are more likely
to benet from the drug. A test that can distinguish people who might benet
from a drug and those who will not benet from a drug is called a biomarker.
Phase 3 studies
Phase 3 clinical trials are large trials designed to denitively prove whether or
not a treatment really works. Phase 3 clinical trials are done to compare a new
treatment to something else, often the treatment that is considered “standard
of care” for that disease and situation. is standard treatment may be another
older treatment or in some cases may not involve active treatment against the
tumor. Phase 3 trials must be randomized; meaning the treatment you receive
is decided randomly by a method determined by the study designers, not by
Chapter 5: Clinical Trials for DIPG
78
79
Chapter 5: Clinical Trials for DIPG
you or your doctors. Random means no one can control what treatment you
get, as if it is determined by the ip of a coin or the roll of a die. e group
you are randomized into is called an arm of the trial. Randomized phase 3 trials
are needed because the results of non-randomized trials, such as many phase 2
studies, can be misleading. Non-randomized trials compare their results to what
one would expect to happen to the usual patient in that situation. However,
non-randomized studies can give a falsely optimistic view of a new drug because:
• People who volunteer for clinical trials are usually healthier and physically
stronger than those who do not volunteer. e inclusion criteria for clinical
trials, as discussed above, weed out people who have other health problems
or are expected to be very sick soon.
• People who can travel to big hospitals for clinical trials tend to have slower
growing cancers than people who cannot travel.
• Over time doctors have gotten better at preventing and managing symptoms
and side eects, so people may live longer.
• People in clinical trials may see their doctor or be contacted by nurses more
often than people not in clinical trials. erefore, problems can be dealt
with early before they become untreatable.
• When doctors and patients think a treatment is working, they are more
likely not to see evidence that it is not working.
Although not very common in oncology, when there is no known eective
treatment for a particular situation, then it is considered ethical and appropriate
to compare a new treatment to giving no anti-tumor treatment. Giving no
anti-tumor treatment does not mean “doing nothing.” It may be called Best
Supportive Care, which means treating all of the symptoms of the tumor but
not giving anything to ght the tumor. Giving no anti-tumor treatment may
also involve a placebo. A placebo is used so that neither the person in the study
nor their doctors know whether the person is getting the new drug that is being
tested. When people and their doctors do not know what treatment someone is
receiving, they are said to be blinded. Placebos prevent doctors and patients from
being subconsciously biased in interpreting MRIs and symptoms by knowing a
person’s treatment arm. Placebos also prevent people from dropping out of the
trial and entering another trial if they are randomly assigned to the arm with
no tumor treatment. A placebo will be a pill or infusion that looks and feels
identical to the active drug, but it does not have any drug in it. Some people
call placebos “sugar pills” because they used to contain sugar instead of drugs.
Many people do not like the idea of getting a placebo or of getting randomized.
Rarely is the outcome without treatment so certain or a treatment so good that
randomized trials are not needed. Some people like to joke that randomized,
placebo-controlled trials are not needed to show that parachutes are helpful if
you jump out of an airplane. However, most cancer treatment situations are
not that clear cut. Although clinical trials are done when it is believed a new
treatment is a good thing, sometimes it turns out that the new treatment is
worse than not treating the tumor as there are unexpected additional side eects.
Some phase 3 trials allow people to receive the experimental treatment from the
other arm of the trial when the tumor grows. is is called crossing-over. For
example, a trial may randomize people between receiving drug "A" or a placebo.
When the tumors of the people receiving the placebo grow, those people may
be able to cross-over to the other arm and receive drug "A". Not all trials allow
crossing over. Whether crossing over is permitted or not depends on the purpose
of the trial, the resources available for the trial, and regulatory requirements.
Side Eects in Clinical Trials
ere is a special vocabulary for talking about side eects in clinical trials. e
side eect of a treatment during clinical trials is called an Adverse Event (AE).
e National Cancer Institute (NCI) has established a standardized way to
measure the seriousness of an adverse event. is is called the Common Toxicity
Criteria for Adverse Events (CTCAE). Over the years these criteria have changed,
and in 2012 the most current version is version 4. e complete CTCAE can
be found at the NCI website: http://ctep.cancer.gov/protocolDevelopment/
electronic_applications/ctc.htm.
Adverse events are graded on a scale from 1 to 5. (Grade 0 refers to not having a
symptom or problem, so someone with grade 0 pain has no pain at all.) Grade
1 adverse events are mild and generally not bothersome. Grade 2 events are
bothersome and may interfere with doing some activities but are not dangerous.
Grade 3 events are serious and interfere with a person’s ability to do basic things
like eat or get dressed. Grade 3 events may also require medical intervention.
Grade 4 events are usually severe enough to require hospitalization. Grade 5
events are fatal.
Most clinical trials and doctors focus on grade 3 or higher events, because those
are the most dangerous. Grade 2 events however, can signicantly impact the
patient’s quality of life, even if they are not medically dangerous. For example, a
grade 1 headache is mild. A grade 2 headache keeps the patient from doing things
Chapter 5: Clinical Trials for DIPG
80
81
Chapter 5: Clinical Trials for DIPG
like shopping or cooking. A grade 3 headache keeps the patient from getting out
of bed even to go to the bathroom.
Consent and Assent
One basic principal of medical ethics is that no person should be included in
any sort of experiment without his or her agreement. is agreement is called
consent. In the past, heinous examples of medical experiments occurred where
people were given injections of experimental drugs or even diseases without
knowing it. However, since the Nuremburg trials in the 1940s and particularly
since the 1970s, experimenting on people without their agreement and consent
has been considered unacceptable in the U.S. and the rest of the civilized world.
Informed consent refers to the idea that not only should people know they
are in a clinical trial, but that they also must understand what will happen to
them during the trial. Informed consent is a process that involves both talking
to someone involved in running the trial to learn about the trial and signing a
paper, called the consent form, that explains the trial. e process of informed
consent should include:
• What is known about the experimental treatment.
• What will happen during the clinical trial, including what medicines will
be taken, when and how they will be taken, and what and when tests or
procedures will be done.
• What parts of the trial are considered standard, i.e., they would happen
even if you are not involved in the trial, and what parts of the trial are
experimental. Experimental parts of the trial can be treatments, oce
visits, tests, etc.
• What the alternative is to being in the trial and what the treatment and
testing would be like if you do not participate in the trial.
• Whether there will be any nancial costs to participate in the trial.
• Whether the trial is expected to benet the participants personally or
whether it is to benet patients in the future.
• Whom to contact if you have questions or complaints about the trial.
• What the procedure is to stop participating in the trial.
All clinical trials are overseen by an Institutional Review Board (IRB), which
is a group of scientists and non-scientists that ensure that clinical trials are done
in an ethical manner. Each university or cancer center has its own institutional
review board. e institutional review board approves all aspects of clinical
trials, including what is included in a consent form. e consent form should
have contact information for the institutional review board in case you ever
feel uncomfortable with what is happening in a clinical trial.
Giving informed consent requires that someone has the mental capacity to
understand his or her options and to make a rational and consistent choice.
Some patients, such as children or people with mental impairments, are
thought to need special protection because they may not understand enough
to give informed consent. In that case, two things are needed. First, the person’s
guardian, such as the parent for a child, must give informed consent. Second,
if possible, the child or impaired person needs to agree to the trial, which is
called giving assent. Sometimes this is impossible, for example for young infants
or people who cannot communicate. e age at which assent is required will
vary from trial to trial, but national groups such as the American Academy of
Pediatrics, and the Children’s Oncology Group recommend that children 7 years
of age or older not be enrolled in clinical trials without their assent. Requiring
assent allows a child to say no and to have some control over what happens
to his or her body. Not only are uncooperative children dicult to get useful
scientic results from, but some children may tire of participating in medical
research before parents, who naturally hope for a miracle.
Questions to Ask
If you are considering participating in a clinical trial, here are some questions
you may want to ask:
1. What phase is this trial?
2. What do we know right now about the treatment being tested and what
is unknown?
3. What is the purpose of the trial?
4. What is the chance that this trial will benet my child?
5. What would my child’s treatment be if he/she does not participate in the
trial?
6. Are there extra tests my child would have to undergo if he/she participates
in the trial?
7. What will I or my insurance be charged for and what will the trial pay for
Chapter 5: Clinical Trials for DIPG
82
83
Chapter 5: Clinical Trials for DIPG
during the trial?
8. If my child get sick or is hospitalized during the trial, who will pay for that?
9. Is the trial randomized?
10. Is there a placebo or blinding, or will I know what treatment my child is
taking?
11. Who is paying for the clinical trial research? Is it the company producing
the drug?
12. Does my child receive any type of nancial reimbursement for participating
in this trial?
Conclusion
All of the advances that have developed to successfully treat many types of
cancers have come from clinical trials. Millions of people are alive today because
of people who participated in clinical trials. However, while clinical trials have
the potential to benet the individual patient and future cancer patients, clinical
trials are scientic experiments and there are important considerations that need
to be understood before patients agree to participate. ere are pros and cons
to participating in clinical trials. A clinical trial may or may not benet the
patients in the trial. Clinical trials limit the exibility of doctors and patients
because it is necessary to get scientically valid results. Asking questions of
your doctor is the best way to get information about any clinical trial you are
considering for your child.
Parent Perspectives
When Caleb’s DIPG was diagnosed, he was less than three months from
his 4th birthday. His tumor was diagnosed in the Emergency Room after an
MRI, and the doctors were crystal clear at the immediate outset that he could
not survive this tumor. Even given this clear-cut, seemingly straightforward
assessment, we decided that a review of our available options was vital.
We were at one of the best Children’s Hospitals in the country; however,
we thought it beneficial to seek other opinions.
Of course, the first thing one does is usually an Internet search. In 2006,
there was considerably less material available for DIPG. Only a few
parents had brought their story to the Internet (we found two); there were
no DIPG support groups, Facebook pages, parent communities, or the like.
The medical literature that was available publicly was clear—long-term
survival with DIPG was unlikely. The parent stories we did find also echoed
that sentiment, and both children had passed away.
We visited one of the premier cancer centers in the US in Houston, Texas, and
found doctors very willing to discuss options with us. They had treatment
protocols for DIPG. I recall the discussion with the doctor vividly: he told
us that long-term survival was not possible. We asked about extension of
life, and he said that this was definitely possible—he could promise a longer
life for Caleb. We asked him what the “extension” would likely be and he
felt certain that “a few months” was possible.
While we were consulting with him, other children were there. One was
vomiting violently from the treatment she had been given. The doctor
shared the details of what Caleb’s treatment would be, and he described the
mouth sores and other side effects the treatment would probably produce.
We were not willing to compromise Caleb’s quality of life for a few extra
months of him being sick from treatment. We wanted what life he had left
to be full of living.
Knowing that Caleb’s long-term survival was not likely, and that treatments
with severe side effects would perhaps extend his life “a few months,” we did
not want to pursue them. Our doctors at the Children’s Hospital provided
us with an option: a Phase II clinical drug study at NIH which was testing
Chapter 5: Clinical Trials for DIPG
84
85
Chapter 5: Clinical Trials for DIPG
the effectiveness of a proposed treatment. Side effects were minimal if any.
We opted to participate in this study, for two reasons: first, it would provide
information to researchers; and second, we did feel like we were doing
something to help Caleb. This treatment, in the end, did not help Caleb.
But it also did not compromise his quality of life.
Caleb survived about ten months past diagnosis. While the initial few
months were challenging with the radiation and Decadron treatments,
his quality of life overall was quite good. He struggled off-and-on with
some neurological problems which were treated with medications, and
his abilities did diminish as the disease progressed. Up until the last few
days before his death, he was alert, aware, and until his last week or so,
he could participate in activities. He was never sick to his stomach, had
just one hospital stay related to the tumor, and was a cheerful little boy.
It’s hard to say, now, that we would have done the same thing as more
treatments are available and some differing degrees of success have been
seen. However, the process would be the same—the compromise to his
quality of life, versus an expected benefit. And, in the end, every parent
has to weigh that carefully for their child and make their own decisions.
Quality of life was absolutely number one from the first day. We told our
doctor that we would NEVER prolong Hope's life without a guarantee
that she would have top quality. We even chose some of our clinical trials
including lack of hair loss as criteria. It was a balance of feeling like we
were doing something to give her a fighting chance (hoping we'd get our
honeymoon and more good days) and not putting her through things that
were for naught.
Lovis just loves to be home. Coming back from a trip, I can see so much what
this means. She really wants to be home. When it comes to trials, yes, we
are interested (of course), but then I couldn't think of taking her somewhere
far away, by plane, when the only thing I hear from her is "When are we
going back home?" I am still stunned by how much travel families do for
and with their children to make treatment possible.
I am still not sure what to do. The inner fight we all know: How much am
I going to compromise my child's quality of life versus a slight chance of
maybe doing something against the tumor with unproven drugs that might
have terrible side effects?
The conversations we always had with her doctors stemmed from the need
to preserve her quality of life—hence no other meds or trials. They would
have provided them to us if we asked, but we both agreed that none of the
approaches were a cure. They helped us understand and accept that there
were trade-offs—her quality of life versus the need to keep her with us
longer. And they leaned very much towards her quality of life.
Emma's case was constantly reviewed. Emma stumped some of the doctors,
because she was on no medications at all—no chemo, no trials—just
radiation and at the end a small dose of dexamethasone to help her swallow.
She survived 16 months. Her doctor said, and I quote, "If we could teach
other parents the approach we had in dealing with our daughter we could
do so much good. We just don't really know how to describe that approach."
I found that interesting to hear, as I don't think we did anything overly
different than what other parents have done—but perhaps we did.
We sent Liam's scans to be reviewed by a leading doctor in the DIPG
community to get her thoughts and opinions going forward. It was agreed
that in fact Liam's tumor had progressed. We also sought the opinion
of another doctor well versed in the care of DIPG patients. We wanted
opinions of studies and clinical trials that might have benefited Liam. We
were told very forthrightly that generally children live six months following
the diagnosis of progression and that oftentimes had little to do with what
new treatments were tried. This weighed heavily on us. We decided (and
were also advised) that what our current hospital could offer Liam for care
would be just as effective as anywhere else. We made the very personal
decision not to pursue a clinical trial at that time in large part because of
Liam’s rapidly declining health. We decided to do a combination of two
chemo drugs in the hopes that they would keep things relatively "quiet" for
as long as possible. Liam by this point was back on higher doses of steroids
and they were increased as needed.
Chapter 5: Clinical Trials for DIPG
86
87
Chapter 5: Clinical Trials for DIPG
During Brendan’s symptom-free periods he regained full abilities and
appeared healthy. He was always doing some sort of treatment though
whether it was off label or clinical trial. His quality of life was very good
aside from the expected side effects of the treatments he endured. He wanted
to fight so he participated in 2 clinical trials—one before progression and
one after. We took our cues from him and as long as he wanted to fight and
live we found treatment options for him.
This decision to not try to prolong her life using treatments was not one we
took lightly, and we are extremely pleased with the high quality of life we
have been able to offer Stella, free of the burdens of invasive medications,
side effects, sedation, etc. that a 2-year old would not have understood.
Because of this decision, we were able to spend the summer taking Stella to
cottages, the zoo, Riverdale Farm, the library and on play-date after play-
date instead of being chained to the hospital. In fact, Stella has been home
the entire time since her diagnosis which has been wonderful for all of us.
Clinical trials were discussed only as a second option if/when the radiation
failed. In hindsight I wish we had gone home and taken a few days to do
some research and consider second opinions. We could have become better
informed and Sam would have had a few days to enjoy his friends while he
was still feeling pretty well. As it turned out Sam never had a "honeymoon"
period so if we had gone home to think and research he could have had a
few days more of "normalcy."
We knew very little about DIPG at that point yet felt like we had to make
critical decisions quickly without the information needed to make informed
decisions. I'm still not convinced that the surgery did anything other than
cause him to forfeit the honeymoon period. There is no way to know if the
surgery did anything to extend his life at that point. Perhaps the radiation
would have stopped the tumor in the cerebellum from growing as it did the
tumor in the pons (at least temporarily). But our goal at the beginning was
to be one of the few to beat the horrible odds and it seemed like surgery
would help that goal.
I wish when Sam was diagnosed that someone could have said, “Here is what
he has, here is the typical timeline, here is our most up to date information
on DIPG, and here are the options including clinical trials. Take it home
and then come back in a few days with questions, and to discuss the path
to take that would be best for Sam.”
At diagnosis there were no choices to be made. If Miguel was going to
have a chance to survive he would need weeks of radiation. My daughter
(Miguel’s mom) spoke with Miguel about his diagnosis and the treatment
he was about to receive. Miguel had just turned eight a month before and
was a very bright child. There really was not an option of keeping things
from him. Miguel’s mom always kept him informed, including showing him
the scans so that he could see for himself.
At progression, it was different. There were choices to make. How do you
know which treatment option to go with when you are given five? My
daughter decided that this decision could not be made alone and that Miguel
would have the final say. There were four primary caregivers for Miguel:
my daughter, her boyfriend, my youngest daughter and myself (Miguel’s
grandmother). My daughter decided that the best approach would be to
analyze and rate each clinical trial individually and then we would come
together to assess the results. The top two options were clear and we all
agreed that there was one that was probably a better approach for Miguel.
Now that there was a choice of two, she presented them to Miguel. It was
important that he was aware of the treatment, including how the medication
would be delivered into his body, and any side effects that were almost
certainly going to occur. The decision was made and the final choice by
all, including Miguel, was Nimotuzumab.
Aimee didn't do any clinical trials; she only did radiation, 6 weeks, 5 times
each week, with external-beam radiation therapy. …She was supposed to
do some new trial that was gadolinium based. She was not eligible for any
trials once she was put on the ventilator….
As parents who knew that radiation was the only thing that had some impact
on DIPG, we were ignorantly devoted to the Phase II clinical trial which
was causing such horrific side effects. We knew that the fact that a therapy
was in Phase II did not mean that it had been effective in Phase I. At the
same time, we mistakenly believed that it would not have moved from Phase
Chapter 5: Clinical Trials for DIPG
88
I to Phase II if it hadn’t shown some promise in the first trial—if children
who received the maximum tolerated dose hadn’t responded positively.
Had we known that the children who received the treatment in Phase I,
even at the maximum dose, did not have a better course than children who
received radiation therapy alone, I don’t know if we would have made a
different choice. In retrospect, I wish we had. If we had had that knowledge
and understood that radiosensitizers were not “new” or “novel,” as we
were told they were, but had been tried in various forms for years, I know
we would have been better informed. We would not be living with the feeling
of regret compounded by guilt as a result of our ignorance.
We had been in a hospice mindset for a few days when we received a phone
call from Andrew’s neuro-oncologist letting us know that a clinical trial
slot (we had previously planned to do) had opened. She knew that we would
most likely choose not to do the trial, but she wanted to allow us to be the
ones to make that decision. Andrew surprised us by saying that he wanted
to participate, so we made a final trip to NIH. An MRI there confirmed
that the tumor had spread throughout the brainstem and to other parts of
the brain. He qualified to do the trial, but when he realized all the pills
that he would need to swallow, he told us that he did not think he could do
it. We were so overwhelmed by the extent of the tumor spread, and by how
well he was doing in light of that, that we could not imagine asking him
to swallow all those pills. Andrew, age 8, and his neuro-oncologist spent
a few moments alone together to talk about the clinical trial and what he
wanted to do. That conversation, between our son and the physician he
loved and trusted, confirmed in our hearts that he was truly ready to stop
treatment. It was clear to all of us that after living life to the fullest for
over 25 months in spite of DIPG, he was tired. He made the decision to
stop treatment understanding exactly what that meant.

Chapter 6
Surgery: What It Can
and Cannot Offer DIPG
Michael H. Handler, MD
e problem with operating on diuse intrinsic pontine gliomas (DIPGs) is
captured immediately by the name itself—they are diuse, they are intrinsic,
and they are in the pons. Tumors elsewhere in the brain tend to grow as a
lump that pushes aside more normal brain tissue, but DIPGs do not. Lumps
of DIPG cells are generally not large individual masses that a surgeon can try
to take out. Instead, the cells making up the tumor project diusely in ngers
that sit widely among other areas of normal brain tissue.
With DIPGs, it isn’t possible to separate normal from abnormal tissue when the
surgeon looks at it. us, an attempt to remove large pieces of tissue to try to
control the tumor (and improve the child’s outcome by getting enough of the
tumor out) is not possible. Instead, attempts to remove abnormal tissue result
in pieces of normal brain tissue being removed. Removal of normal tissue can
also happen during surgery on other parts of the brain, but in other parts of
the brain the nearby normal tissue usually doesn’t have as important a function
as pons tissue.
e Pons
e pons is a small area of the brain about 3.5 cm. long and 2.5 cm. wide.
In it are the brain centers that control sleeping and waking, eye movements,
facial movement, and hearing. e major pathways for movement of the arms
and legs, and for most of the body’s sensations, pass through it in a complex
manner (see chapter 3). e cerebellum
which sits behind the pons sends bers
from one side of the brain to the other
through the pons. e cerebellum controls
smooth muscular movements and balance,
Dr. Handler is a Professor and
Chairman of Pediatric Neurosurgery
at the Children’s Hospital Colorado
and the University of Colorado
School of Medicine, Aurora, CO.
89
Chapter 6: Surgery
and some of the balance centers of the brain are located in the pons. Additional
centers controlling balance are located in the medulla. us, the pons is a
critical structure that highly inuences the entire body’s ability to function
properly. When the pons is damaged, it can have an extremely wide impact on
the body’s ability to sustain itself. Any surgeon should be extremely cautious
when attempting an operation in this region. Taking small amounts of tissue
may be possible without inicting serious neurologic damage, but taking large
amounts of tissue simply is not.
History of DIPG Diagnosis and Imaging
Surgeons’ current thinking about DIPG tumors is based on the tumors’ history.
In the past, surgeons would generally try to remove brain tumors. But that was,
and is still, rarely possible in the pons. With tumors in other areas of the body,
surgeon’s usually try to take a small amount of tissue with a “needle biopsy”
to gain a better understanding about the tumor before considering a larger
operation. Biopsies, however, were rarely done on brain tumors, because biopsies
were much more dicult in the brain than elsewhere in the body. e reason for
the diculty was that it used to be very dicult to know exactly where in the
skull, and therefore where in the brain, a needle should go to make a diagnosis.
While surgeons used a variety of external landmarks for placing needles in the
brain, this technique—called stereotactic biopsy—was not good enough for
widespread use. Forty years ago, that began to change. New computerized
brain imaging technology suddenly made it possible to see anatomic details in
a remarkable way. e technique of stereotactic biopsy became feasible, and
surgeons began to use it.
Meanwhile, brain imaging itself became much more precise. In particular, MRI
revolutionized surgeons’ ability to understand and visualize the location of
tumors, particularly in the posterior fossa—the back of the brain where the pons
is located. MRI and stereotactic technology were now linked in a constructive
way, and surgeons began to try stereotactic biopsy in the pons.
Clinical Study Recommendations for Standard of Care
Pediatric oncologists have long worked collaboratively to try and develop the
best new treatments for tumors, in the most ecient way. One such collaborative
study, organized through the Children’s Cancer Group (the CCG study 9928),
looked at patients with diuse pontine gliomas and proposed new treatments.
is study, published in 1993, concluded that when an MRI shows specic
characteristic features of changes in the pons, it was sucient evidence to make
a DIPG diagnosis without a biopsy. It also turned out that when a biopsy was
taken, the result of the pathology was not helpful in planning a particular
treatment strategy and did not appear to alter a child’s subsequent outcome.
erefore, the recommendation became that no biopsy should be undertaken
in cases that appear to be typical pontine gliomas.
At that time, the risk for harm from a biopsy was not yet well known, because
the technology for stereotactic biopsy was still relatively new. But because the
benet from biopsy is so small, the risks were too large for it to be undertaken.
Based on this historical data, the accepted standard of care has become that no
biopsy need be done for DIPGs.
When the neurosurgeon, oncologist, and radiologist agree that a tumor does
not appear to be a typical DIPG it is considered appropriate to proceed with a
biopsy. Since the publication of the CCG study, many more publications have
documented the safety of stereotactic biopsy in the posterior fossa with relatively
little inicted harm. us, the decision to do a biopsy is based on whether the
tumor appears enough atypical that a dierent diagnosis could be considered.
In addition, some tumors—in the opinion of the surgeon, oncologist, and
radiologist—remain so unusual that an open operation (removal of a ap of
the skull and entering the brain tissue with instruments larger than a needle)
is more appropriate.
Stereotactic Biopsy
A stereotactic biopsy is based on obtaining a computerized image on which
a target (tumor) can be identied; that image can be an MRI or CT scan.
Technology is then used to calculate the position of the target in relation to the
scan. Several dierent technologies are available to do that, some using a rigid
frame that is attached to the head before the scan, and others that can map the
surface of the face and use that as the basis for determining the tumor’s position.
With the head in a xed position and the trajectory calculated, a very small hole
is made in the skull to allow the needle to pass to the target and obtain the tissue.
e tissue is then removed and prepared so as to identify the pathology; other
studies may also be done. Stereotactic biopsy has the advantage of a very small
incision, a small hole in the bone, and less risk than a larger open operation.
Most studies of stereotactic biopsy have shown relatively little damage as a result
and a very low rate of death from the procedure (less than 1%).
Chapter 6: Surgery
90
91
Chapter 6: Surgery
Deciding When to Operate
e decision whether or not to operate must be based on the imaging features of
the tumor and on whether the treatment is appropriate for the individual child.
e DIPG’s location presents a problem (in addition to that of the tumor itself).
e brain produces cerebrospinal uid (CSF), which circulates through a series
of spaces in the brain called the ventricles, which include:
• Lateral ventricles: One lateral ventricle is found in each cerebral
hemisphere, and each lateral ventricle communicates through a small
channel (called the foramen of Monro) to the third ventricle.
• ird ventricle: e third ventricle sits on the midline at the base of the
brain and ends in a channel, called the aqueduct of Silvius that goes to the
fourth ventricle.
• Fourth ventricle: e fourth ventricle sits behind the pons and medulla,
in front of the cerebellum.
CSF goes downstream from the lateral ventricles to the third and fourth,
then leaves the fourth ventricle through channels that connect to the space
around the brain (the subarachnoid space) where CSF is absorbed into the
bloodstream. Because DIPGs sit adjacent to this uid pathway, as they grow
they may at times block the ow of CSF. is blockage causes uid pressure
in the brain to go up and the uid spaces to enlarge—a condition called
hydrocephalus. When the uid pressure builds up, it can cause headaches,
vomiting, mental changes, and even coma. us, sometimes children with
DIPGs need procedures to control the uid pressure.
Treating Hydrocephalus
e most common and well-established procedure to treat hydrocephalus is the
ventriculoperitoneal shunt (VP shunt). A tube is placed from the outer surface
of the head through the skull and brain into a lateral ventricle. e shunt is
connected to a device—the valve—which determines how much pressure must
build up before CSF starts to ow, and makes sure CSF only ows out of the
ventricle and not back in. e valve sits under the skin and connects to a tube
that leads to a place in the body where the CSF can be absorbed back into the
blood stream (where it would have gone from the brain directly, if it could).
e most common end point of shunts is the peritoneal cavity—the belly—but
other times shunts go to the chest, through blood vessels to the heart, or even
to the gall bladder. Eective shunts have been around for about 50 years, and
they save and improve the lives of tens of thousands of kids each year.
Shunts, however, have their own problems. ey are foreign to the body, so
the body may react to them and block o their ow. ey are also mechanical
systems that can break or malfunction. e most common way they become
a problem is when they become infected. Infections happen between 5 to 14
percent of the times they are implanted. (Neurosurgeons are very focused on
how to reduce the rate of these infections.) When a shunt is infected, it has to
be removed, and—after a period of time—replaced with a clean system.
Because of the problems with shunts, another way to drain uid from the
ventricles is the endoscopic third ventriculostomy (ETV), which creates an
alternative pathway from the third or fourth ventricle when the normal pathway
is blocked. Whether the ETV is an option depends on the particular anatomy of
a child with hydrocephalus due to a DIPG. is is a matter only the surgeon can
assess. (e pons sits behind the area where CSF goes after an ETV. If a DIPG
can block the CSF pathway behind it, it can block the pathway in front as well.)
Biopsy of More Routine Tumors: Why or Why Not?
A fundamental ethical dilemma raised by the current standard of care is that
when the tumor looks typical on MRI, the recommendation (based on research
completed 20 years ago) is that no biopsy should be done. e potential risk
for the individual child, as interpreted at that time, could not be balanced by
the potential benet. Since then, two things have changed:
1. Many more patients have undergone safe biopsy of brainstem masses than
had done so when that observation was made;
2. e ability to study tumors has been vastly advanced by genomic analysis.
DIPG tumors will not be understood as other pediatric brain tumors are,
unless more tissue is obtained for study by contemporary techniques. e
cancer community will have to decide the most appropriate course of action
with DIPG tumors.
Federal Guidelines for Clinical Research
Federal guidelines for conducting research, particularly in children, stipulate
that there must be the potential for benet for the individual child if he or she
is to undergo a procedure with more than a minimal amount of risk. What
will be the benet to undergoing a procedure; will the procedure allow us to
better understand the nature of the tumor; and will it benet a particular child?
How should we measure benet? If the child and his or her family decide that
Chapter 6: Surgery
92
93
Chapter 6: Surgery
to help advance knowledge is a very signicant benet to that child, is that
substantial enough to oset the notion that because “the child’s survival won’t
change,” there is no potential benet? is is the overarching question that has
no easy answers.
Parent Perspectives
Our son Caleb, had his DIPG diagnosed at a critical point where his tumor
had grown to the point of obstructing cerebrospinal uid (CSF) circulation be-
tween the brain and the spinal cord. The higher ventricular pressure, from the
CSF obstruction and resulting hydrocephalus, was visible on the MRI and a
syrinx (pocket of uid) was also evident. Clinically, Caleb was sluggish, limp,
couldn’t keep his eyes open, and was clearly having problems from the hydro-
cephalus. So we had an immediate problem which needed to be addressed.
His medical team recommended a ventriculoperitoneal (VP) shunt be placed
immediately.
We struggled with the decision of what to do. This was “Day 1” for us. The
idea of DIPG, shunts, radiation, what we would do to treat Caleb—these
things were all questions we faced immediately. The shunt issue, however,
seemed to be the most pressing since the hydrocephalus needed to be promptly
addressed. We needed time to gure out what to do about the base DIPG diag-
nosis, and the shunt bought us that time.
Nonetheless, the idea of operating on Caleb’s brain was not an attractive one.
We assembled his medical team in the room, and the question we put directly
to them was, “If this was your son, what would you do?” With zero hesitation
the neurosurgeon quickly said, “I’d place the shunt.” In hindsight, this was
one of the most simple and straightforward procedures performed on Caleb.
After the surgery, Caleb was awake and ready to play. His body adjusted to the
shunt almost immediately.
Caleb’s VP shunt worked well. We never had problems with it. As the tumor
progressed, he did have other neurological problems which occasionally
brought him to the emergency room—and the very rst thing the doctors would
do was check the operation of the shunt.
In hindsight, we would do that part of Caleb’s treatment the same way again.
If his DIPG had been diagnosed at an earlier stage when hydrocephalus was
not already a problem, it might have been a different question for us. However,
given the point we were at, the shunt was the treatment that bought us the time
to treat him with radiation and consider our other options.
Chapter 6: Surgery
94
95
Chapter 6: Surgery
Quickly our attention became focused on what we needed to do next to help
Liam. Liam's neurosurgeon reviewed with us the most pressing issue at the
moment which was the uid that had accumulated on his brain. We opted for
a 3rd Ventriculostomy. Not every child is a candidate for this surgery nor is
it performed by every doctor. The signicant risk was explained to us. We felt
extremely condent in our doctor and felt this would be the best option for
Liam going forward.
After this decision was made we walked the long hallway back to Liam’s room
to give him the news. Not ALL of the news mind you, but a small piece and the
next steps that needed to take place. The three of us sat with Liam, told him
that his doctor needed to help him feel better and that there was too much uid
in his brain that shouldn't be there and he would need surgery. We explained
that he would be asleep and wouldn't feel anything and would wake up and be
able to see Mom and Dad right away—which the operating room staff made
happen for us. At this point in time, Liam was so sick and weak he barely re-
sponded but simply nodded his understanding. Our sweet boy looked every bit
of his little six year old self in that hospital bed and our hearts were absolutely
broken but also incredibly resolved to do everything we needed to protect and
help him on this journey.
As the weeks and then months went on, and with the help of a successful 3rd
Ventriculostomy, surgery, effective radiation treatment, and the use of steroids,
Liam's symptoms greatly improved. He returned to school and eventually came
completely off steroids and became symptom free. His meds and periodic vis-
its to his oncologist were just a minor interruption to the real business of the
day—being a kid.
Our experience with neurosurgery was brief, I suppose. The neurosurgeon put
in Tatumn's shunt at diagnosis since she did have some hydrocephalus. It did
relieve the pressure but it was always an uncomfortable question to ask our-
selves, "so how long will this thing be in her head?" when we knew the answer
wasn't relevant given the prognosis.
After Tatumn's six week run of radiation we started to see signs of the “hon-
eymoon period,” and we were reducing her steroids. We saw some light at the
end of the tunnel! Tatumn was using her hands again and was speaking better
and we had hopes of her staring to walk again. Well, high fevers set in and she
started to be very uncomfortable to move. We had to be slow to move her and
she had denite abdominal discomfort. They checked her blood for infection
but couldn't nd anything. They did an x-ray and didn't see anything either.
Our neuro-oncologist wasn't available. It was during Thanksgiving week so
the doctors we saw said maybe it was the tumor and her body temperature
wasn't being regulated right. But I could sense it was more. So we went on
her wish trip and fevers kept spiking. I called her radiation oncologist when
we got back and he said to take her in immediately and have her shunt tested
for infection. He was right. Tatumn had an infection probably caused by the
shunt and since it emptied into her peritoneal area I think that is why she had
the discomfort. The shunt was removed and during our 10 day hospital stay on
antibiotics (right before Christmas) they monitored her cerebral uid and said
she would be ne without it and they didn't put a new one in. We felt so good
to have that gone.
What do I wish I had done differently? I wish I had listened to my intuition
more. I knew Tatumn's fevers were more than tumor related. Maybe we would
have caught it sooner and her wish trip would have been better.
About a year after diagnosis, our son began to show symptoms of tumor pro-
gression. It was at this point that we chose to meet with a pediatric neurosur-
geon to explore the possibility of surgical intervention, if necessary, to treat
hydrocephalus. We were more interested in an ETV than a shunt because it
seemed less invasive to us. The neurosurgeon conrmed what we had heard
from others—that surgical intervention to treat hydrocephalus is not recom-
mended for most patients with DIPG. While surgical intervention may prolong
life, and even help with quality of life in some ways, it does not stop the tumor
from growing and affecting the body in other ways. So it's possible to treat
hydrocephalus successfully and prolong the child's suffering.
As I understand it, many of Bizzie's initial symptoms were related to hydro-
cephalus. We were told that she needed surgery; it was not presented as a
choice of having surgery or not, but rather ETV or shunt.
This was presented
to us along with her diagnosis, so we were at the hospital, in the PICU,
Chapter 6: Surgery
96
97
Chapter 6: Surgery
di
gesting the diagnosis. In fact, the pediatric neurosurgeon was the one who
ofcially diagnosed Bizzie. We were still so shell shocked when we made the
decision. We chose the ETV surgery. We did so based on the facts as they were
presented to us. My husband also spoke with another neurosurgeon so that we
had a second opinion about the surgery. We decided that we should go ahead
with the ETV and then consider moving to a large cancer center for treatment.
I don't think we understood, though, how important the facility and nursing
staff is in recovery.
We were admitted on Friday and Bizzie's surgery was scheduled for Monday.
We were given the impression that Bizzie's level of hydrocephalus presented
an emergent situation, but that they also wanted the steroids (dexamethasone)
to be given a chance to do some work. Of course, it was also Friday. Monday
morning came and it was time for Bizzie's surgery. I was sick. I was shaking
holding her as the nurses and transport fought over stupid logistics. When I
sputtered out a plea for help, the nurse looked at me and said in a testy voice
"What? I can't understand a word you say." That memory still sticks with me.
I wasn't looking for sympathy, but I really needed some help getting through
that moment.
Bizzie refused to take off her princess dress and wear the standard green gown.
My husband and I begged and pleaded. Finally, we convinced her that it was a
Tinkerbell costume because it was green. I rode down on the bed with Bizzie in
my lap. They sedated her and the surgery began. The surgery was successful.
She came back up to the PICU for recovery. In the early morning following
her surgery, Bizzie was transported by the PICU nurse, the receptionist, and a
technician to the MRI area. My husband was with them while I tried to sleep.
When they returned, Bizzie had a seizure and they intubated her. The doctors
went back and forth as to the cause—low sodium levels, or the monitoring
piece near her brain perhaps brushed something in transport. Bizzie remained
on the vent for two days. They told us that she would not be able to breathe
on her own when they pulled the tube, but she did. She did not talk for several
days after that. She just stared. Our PICU neighbor thought that Bizzie had a
twin—the girl she saw before the surgery who was making quite a racket in the
PICU, and this other girl who did not respond to anything. Bizzie did recover
slowly, although she didn't walk again for several months. But that was the
worst she was until the day before she died.
Sam's tumor extended into the cerebellum and the neurosurgeon recommended
we do surgery to remove what he could from that area. I was under two mis-
conceptions at the beginning—one that the tumor in the cerebellum was a fo-
cal rather than diffuse tumor and second that diffuse was more like tentacles.
Later I learned that that the cerebellum tumor was also diffuse and that dif-
fuse was more like sporadic tumor cells spread throughout, and not really
connected like you would think of a tentacle being. I don't know that either
misconception affected our decision to do surgery but I wish I had had a better
understanding in the beginning nonetheless.
I know most kids with DIPG never go through surgery like this but perhaps
Sam's experience will be informative anyway. The surgery itself lasted 4 to 5
hours and was terrifying to think of him in there where anything could happen.
Everything went smoothly though, and he actually recovered well in the rst
few days. They had “PT, OT, and Speech” evaluate him and it was decided he
could handle outpatient rehab as opposed to inpatient. We were so excited to
have him come home. At that point he needed a little help walking but other-
wise seemed ok. That was on a Wednesday. When he was released I went over
the prescriptions he would need at home, specically asking the resident dis-
charging him if he needed to still be on the steroids and was told that he didn't
need them anymore. What a fool I was to accept that answer.
The next few days Sam seemed ne. There was nothing unusual, considering
his surgery. Then by Sunday he seemed to be having more trouble walking and
was a lot more tired. He'd had a lot of friends visiting so we thought it was
from trying to do too much too soon. Monday night he threw up and Tuesday
he could barely walk so we headed off to the emergency room where they did
an MRI and said the tumor was reacting to the surgery. The neurosurgeon said
the area in the cerebellum was ne but that the tumor in the brainstem was
causing problems. They bumped up the schedule for radiation and gave him
lots of steroids. By now he could not even sit on the side of the bed without
help, let alone walk; his right eye was turned inward causing double vision; he
had lost his hearing in his right ear; and he had difculty swallowing. He was
released to inpatient rehab for 10 days then home again. He started on the 6
week course of radiation and the Temodar while in rehab. When he came home
from rehabilitation he was so much worse physically than when he came home
from the surgery. It broke my heart to think that it could have been prevented
had he been on steroids at home.
Sam spent the next three months working so hard with his therapists to get bet
ter.
Chapter 6: Surgery
98
99
Chapter 6: Surgery
By June he was walking with a cane and able to do much more for himself
again. He was even on track to get prism glasses for his double vision. He
stayed pretty stable from mid-June until mid-August then had problems with
his balance. Within a matter of days he could not walk again—this time from
the balance issues. So off to the emergency room again where they determined
he had a cyst growing in the cavity in the cerebellum where they had removed
the tumor before. It was pressing on the fourth ventricle causing his balance
problems. So he had another surgery to remove that and came home again—
this time with steroids. Unfortunately, the tumor in the brainstem seemed to
have been aggravated because he never bounced back from that surgery and
in fact on his next MRI there was progression.
In hindsight knowing what I know now I might have pushed for no surgery
and just gone with radiation and some other chemo. I think he may have had a
longer time until progression and a better quality of life during that time. But
there is no way to know. Others think that without the surgery he may have had
less time.
Chapter 6: Surgery
100

101
Chapter 7: Radiation Therapy
Chapter 7
Radiation Therapy
Arthur Liu, MD, PhD
Radiation therapy is the standard treatment for children with diuse intrinsic
pontine gliomas (DIPGs). is type of therapy uses high-energy x-rays, similar to
those used in a computed tomography (CT) scanner but at much higher doses.
ese x-rays deposit energy within the tumor, causing damage to the DNA of
cells. e tumor cells are then unable to repair the damage, and ultimately die
when the tumor cells try to divide.
e Radiation erapy Process
Before starting radiation, parents of a child with a DIPG consult with the radiation
oncologist to discuss the planned therapy and potential side eects. After this initial
consultation, the child undergoes a planning session (also referred to as a simulation).
At that time, a mask will be custom-made for the child. e mask allows accurate,
consistent positioning of the head for each treatment and helps the child remain still
during treatment. e mask is made out of a special type of plastic that becomes
moldable when heated in a water bath. After the mask is completed, a special CT
scan is performed. is entire process takes approximately 1 hour.
After the planning session, the radiation oncologist uses the CT scan to dene the
area that corresponds to the tumor and the regions of the brain that should not receive
radiation. With the assistance of dosimetrists (who specialize in calculating the dose
of radiation to ensure the tumor gets enough radiation) and physicists (who develop
and direct quality-control programs for radiation equipment and procedures), a
radiation therapy plan is developed to maximally treat the tumor while minimizing
the amount of radiation delivered to normal brain tissues and surrounding tissues.
Radiation therapy is then delivered daily,
Monday through Friday, for about 6 weeks to
a total dose of about 54 Gray (Gy). Smaller
daily fractions accumulating to the total dose
over weeks is intended to allow normal tissues
the chance to repair some radiation-induced
Dr. Liu is the Director of
Pediatric Radiation Oncology
and Program Director of the
Radiation Oncology Residency
Program at the University of
Colorado, Denver, CO.
Chapter 7: Radiation Therapy
102
103
Chapter 7: Radiation Therapy
DNA damage while still destroying the tumor.
As radiation patients must lie still and alone on a table, some children are too young
or too ill to tolerate the radiation treatments while awake. In these cases, the planning
session and treatments can be performed under general anesthesia. A commonly
used anesthetic agent in radiation therapy is propofol. Propofol is an intravenous
anesthetic that allows for rapid induction and recovery, and—most importantly—
does not require intubation (insertion of a tube) for protection of the airway. Even
with daily use, the risks of complications with propofol are very low. Typically, the
child is brought into the room awake, with the parents, and the anesthesia is initiated.
After induction of anesthesia, the parents leave the room and the radiation therapy
procedures are performed.
Eectiveness of Radiation erapy
Radiation therapy is an eective palliative treatment that improves symptoms
in about 80% of children with DIPGs. e dose of radiation therapy is limited
by the tolerance of the surrounding normal brain tissue. However, given the
high rate of response, in the 1980’s a number of institutions increased the dose
of radiation therapy from 54 Gy to greater than 70 Gy and had promising
preliminary results. To escalate the dose of radiation delivered, smaller doses
per treatment were used (referred to as hyperfractionation). ese smaller doses
allow greater recovery of normal tissues, and thus a higher total dose can be
given. However, a Pediatric Oncology Group randomized trial showed that
while the higher dose of radiation was well tolerated by most children, there
was unfortunately no dierence in survival rates.
Medical professionals have an interest in exploring agents that may be given
along with radiation to improve the eects of this therapy; these agents are
referred to as radiosensitizers. Some types of chemotherapy, such as carboplatin,
can be used as radiosensitizers. Other experimental agents are also being studied.
For example, arsenic trioxide given concurrently with radiation therapy is
undergoing clinical trials to determine safety and to provide information that
can be used for studies of eectiveness.
Possible Complications of Radiation erapy
A common complication of radiation therapy in children with a DIPG is
radiation necrosis—cell death of brain tissue. is may cause swelling and
potentially lead to neurologic symptoms such as headache, nausea, vomiting,
cranial neuropathies, and ataxia (loss of muscle movement coordination).
Radiation necrosis can be very dicult to distinguish from tumor recurrence
by manifesting clinical symptoms or by imaging. Steroids are typically used
for the symptomatic treatment of radiation necrosis. However, steroids can
also cause side eects, including behavioral issues, insomnia, and weight gain.
ese steroid-related complications can signicantly impact the child’s quality
of life. e exact mechanism of radiation necrosis is poorly understood, but
vascular endothelial growth factor (VEGF) appears to play a role. Bevacizumab
is a monoclonal antibody that interferes with VEGF and is being studied as a
possible treatment for radiation necrosis.
Technologies for Delivering Radiation erapy
Many dierent technologies are used to deliver radiation therapy. e most
common radiation therapy machine is a linear accelerator, in which high-
energy electrons impact a target to generate high-energy x-rays. ere are a
number of dierent manufacturers, but most of the machines only have slight
technical dierences. Some machines provide the ability to perform a CT
scan for localization; these machines include Tomotherapy (Tomoerapy
Incorporated, Madison, WI), Trilogy (Varian Medical Systems, Inc., Palo Alto,
CA), and Synergy (Elekta, Stockholm, Sweden). e Novalis Tx (BrainLAB,
Westchester, IL) uses orthogonal planar x-ray imaging for localization. ere is
no clinical dierence in any of these machines. From a technical standpoint, the
Cyberknife (Accuray, Sunnyvale, CA) is the most dierent from other machines.
e Cyberknife mounts a linear accelerator on a robotic arm and is primarily
used to treat small tumors throughout the body. Due to the relatively large
size of the brainstem tumor in children with DIPGs, Cyberknife is typically
not an option.
Proton radiation therapy (PRT) is a form of radiation therapy that has very
limited availability. PRT uses protons to deliver therapeutic radiation. Protons
dier signicantly from the photons used in conventional radiation therapy
because they have no mass or charge, compared to protons, which have mass and
are positively charged. e mass and charge of protons results in a phenomenon
called the Bragg Peak, which results in no energy deposited after a certain depth
in tissue depending on the energy of the proton (higher energies go deeper). is
technique allows protons to potentially deliver less radiation therapy to normal
tissues, with fewer late eects of therapy. Numerous theoretical modeling studies
have shown benet to using proton radiation. For children with DIPGs, the
potential benet of protons is unfortunately minimal. e dierence in normal
tissue radiated between PRT and current photon radiation therapy techniques
Chapter 7: Radiation Therapy
104
105
Chapter 7: Radiation Therapy
is small, and late eects in these children is not yet a signicant concern due
to the extremely low survival rate in this population.
In summary, radiation therapy is the current standard treatment for children
with a DIPG. Radiation therapy improves clinical symptoms in the majority of
children, but that improvement is temporary. e most active areas of research
are exploring the addition of therapeutic agents to a backbone of standard
radiation therapy.
Parent Perspectives
Radiation is the ONLY thing that has proven to help SOME of our kids so
we chose to do it. I don't regret it necessarily as we were fighting for our
child. The steroids took way more than the radiation did. While our daughter
didn't get the “honeymoon period” that many children do, we continued
to believe that we would be the recipient of a miracle. She only lost some
hair in the back but no one knew it because her hair was long enough to
cover the loss. She did not lose her eye lashes.
Andrew's pediatric oncologist called the tumor a pontine glioma. She said
that the standard treatment was radiation, and that it could possibly shrink
the tumor temporarily. She told us that sometimes people use chemotherapy
in an attempt to make the radiation more effective. Andrew started radiation
with two chemotherapies, but we quickly chose to stop that part of the
treatment. We decided that the possible benefit of the chemotherapies was
not worth the definite side effects (nerve pain, nausea, etc.) Radiation alone
was the right choice for us. Andrew's tumor did not begin to grow again
until a year from diagnosis.
Hope got no “honeymoon.” Hope saw no relief from the radiation. We
were devastated as her tumor barely shrunk at all. It began to grow almost
immediately and had areas of necrosis which caused further harm to our
beautiful girl. Hope was courageous. She continued to speak in terms of,
“when I get better” and “after treatment.” She attended therapy even when
it hurt because she knew it was “good for her.”
The first time our son had radiation as an outpatient, we arrived at the
hospital by 8:00 a.m. The nurse and aide checked him over, hooked him up
to I.V. fluids and monitors, and took him down for sedation and radiation
at 9:00 a.m. A nurse changed the dressing for his port while he was still
sedated. Another nurse monitored his vital signs while he recovered and
made sure he was able to drink, eat, and use the restroom. We didn't get
Chapter 7: Radiation Therapy
106
107
Chapter 7: Radiation Therapy
home until noon. I had not expected to be there for the whole morning.
The radiation treatment takes moments, but the preparation and recovery
take more time.
He began to lose some hair two or three weeks into radiation. It looked
like someone had taken clippers and shaved off one to two inches around
the outline of his ears. We had his hair cut short so that the hair loss was
not as obvious.
We were able to get a second round of radiation. When we first talked to
our neuro-oncologist about re-irradiation she said it simply was not an
option. A couple of weeks later I spoke to her about the work being done
at another treatment center and she was mildly accepting of it; then when
I presented her with the ISPNO abstract she began to consider the idea
and was willing to look into it further. Bringing concrete information to
her definitely helped.
If parents are interested I would suggest starting to talk about re-irradiation
early. We had been told radiation was a one-time deal. I found that talking
about it each time I saw his doctor was helpful. At first she said, “No way,
absolutely not." As time went on and I presented her with more information
she became more open. By the time progression happened she knew we
wanted re-irradiation and went to bat for us with the radiation oncologist.
The radiation oncologist who did the first round of radiation decided she
would not do it so our doctor found another that was willing. The radiation
oncologist that did the second round said that the swaying factor was that
she knew that we knew it was not a cure. She knew we were looking for a
second “honeymoon period” and she felt that was reasonable.
My nephew had an experimental new radiation course that a radiologist
was studying. He had just five days of a higher dose of radiation instead
of 30 days at a lower level.
I spent days and nights on the computer and on the phone with numerous
doctors; we sent Ellie’s scans everywhere. We were given the typical
radiation/chemo option knowing very well radiation could not be put off
for long. We also learned that jumping into a treatment option may preclude
you from others. While with this monster there is no right or wrong we
wanted to receive a good feel for all available options. We explored both
proton beam radiation and IMRT (intensity-modulated radiation therapy).
After consulting with two neuro-oncologists we opted for IMRT and were
very happy with this decision.
During Ellie’s six week IMRT we were determined to pinpoint what next
steps we would take. Ellie tolerated her radiation therapy in very good
health. She really did very well. She never had to be sedated and with the
exception of fatigue, experienced little side effects. Her determination to
gain her strength back and get back to all the activities she sorely missed,
served her well.
We met with the radiation oncologist, who explained to us, and to Bryce,
about what radiation would be like. Bryce had another MRI to plan his
treatment. Then we went home to face our family and friends after being in
hospital for 5 days. We were home for one week, and Bryce chose to go to
school, to keep things normal as much as possible. I think back on that now,
and even that is telling of his character and how Bryce handled all of this.
I remember asking his doctor how long Bryce would have had without
treatment, and was given the answer of one month. These radiation
treatments would give us time to learn to live with this new reality, and
hopefully the treatment would make him feel better because he was already
having headaches and dizziness, as well as having difficulties with poor gait.
So, from March 10th to April 23rd, we stayed at Ronald McDonald House
from Monday to Friday as Bryce underwent radiation. We made another
good decision—to keep our whole family together during treatment. By the
end of it, Bryce was 50 pounds heavier from steroids and had lost his hair,
but the earlier dizziness and headaches had gone away for the most part.
I have to tell you that when treatment was over, I was scared to death. So
what were we supposed to do now, go home to wait for him to die? We
were so happy to be going home, and yet terrified, because we knew that
at some point, Bryce would lose his battle—the tumor would resume its
growth. Bryce deserved to go home and just be 13—not go home to wait
for the other shoe to fall.
Chapter 7: Radiation Therapy
108
109
Chapter 7: Radiation Therapy
We chose to do radiation treatment concurrently with Temodar® and the
tumor shrank considerably. We stopped chemotherapy after much prayer
and used a nutritionist to fully change Kayla’s diet. We used many, many
alternative therapies and her tumor shrank further. We were incredibly
blessed with 4 ½ years of generally symptom-free living for Kayla. She
was in piano, ballet, and did well in school. When she was 10, her cancer
returned and she died 5 months later on June 18, 2010.
Courtney was admitted to the hospital and started on steroids. We were given
the option of the standard chemo/radiation treatment. We considered taking
her to a well-known children’s hospital or somewhere else for treatment,
but decided that since they couldn't offer us anything that would give her a
better chance of survival we would stay close to home. We were confident
that she would get as good of care at our local children's hospital as she
would anywhere else.
She had 6 weeks of radiation and about 5 weeks into it she started having
more muscle weakness in her legs. By the time she had completed radiation
she was in a wheelchair. Courtney was on Avastin®/Temodar® after she
completed radiation. I wish we had just given her the radiation treatments
and left off the chemo. The Temodar® wiped out her white blood cell counts
and because of this it really affected her quality of life. She had to spend
114 of the 186 days she lived after diagnosis in the hospital. She was taken
off the chemo in August because of this.
October 6th we had to take her to the hospital because she had a severe
headache. An emergency MRI was done and it showed the tumor was
progressing in the spinal area. At this point there was nothing else we
could do. She couldn't start on chemo because her white count still had not
recovered and she was ineligible for re-irradiation because it had only been
4 months since she had completed her first round. We took her home and
planned to cherish the time we had left with her. Three days later she had
to be admitted to the hospital because she was having difficulty breathing
and severe headaches that we couldn't get under control. She spent her last
22 days of life there and died on October 31, 2010.
After quite some pushing from our side, radiation started and was successful.
The doctors still felt uncomfortable sedating her but I remember thinking
"If she is going to die because of this tumor right now, we might at least
try radiation. I wouldn't blame anyone if the sedation did go wrong.” At
the same time I was wondering how I was able to think that without losing
my mind.
Liam endured the standard six week protocol of radiation along with a bolus
not always done at the end of that standard time. We were told radiation was
the only thing known for this kind of cancer that truly had the possibility of
shrinking the mass. DIPG was much like sand sprinkled in Jello we were
told. Radiation was very effective. Liam's tumor shrank considerably.
Stella is not, and has not, received any treatment. Because Stella was barely
2 years old at the time of her diagnosis, the standard treatment of radiation
therapy would have entailed 6 weeks of Stella being sedated on a daily basis
to receive the therapy. With no guarantee it would work, Stella's young age,
and the fact we wanted desperately to enjoy our remaining time with her, we
opted for no treatments. She was on steroids for one week post-diagnosis,
but we despised the changes we saw in her (huge appetite, tantrums known
as "roid-rage," discomfort, etc.) so we took her off immediately with no
plans to put her back on.
We did the proton radiation at a facility near where we live. I was told that
there would be fewer side effects than regular radiation. Warren started
off doing fine. He only lost a small amount of hair, and you really couldn’t
tell. He was tired but not overly tired. But at the same time he was doing
radiation he was also doing physiotherapy, so that played a role in him
being tired as well.
Joseph responded well to radiation. Our neuro-oncologist told us that the
first post treatment MRI showed remarkable tumor reduction however,
his clinical response was poor even after the second MRI showed more
reduction in tumor size.
Chapter 7: Radiation Therapy
110
The radiation-oncologist told us to expect fatigue. He slept twenty-two
hours a day. After our second visit to the neuro-oncologist after radiation
therapy they called it somnolence and increased the dexamethasone which
kept him awake twenty-two hours a day and totally changed his personality.
I remember looking at the MRI wanting to find something solid not a white
cloudy section. It was never truly explained to me what size a normal pons
should be or why my boy did not respond to treatments after evidence
of radiological improvement. We had set up second and third opinion
appointments but Joe did not make it to them.
I wish I had known that not all DIPGs respond to radiation. I would not
have waited for him to improve clinically to do more activities together
as a family.
When my 17 year old brother Daniel was diagnosed with DIPG, our family
was very overwhelmed. The doctors tried very hard to get my brother to
begin radiation immediately and this worried my parents very much. As
Daniel's oldest sibling, (and not living at home for many years) I was close
enough but not too close to be paralyzed by the fear and pressure from the
doctors. I realized that it would be wise to devote a few days to get second
opinions, and research all the options and make an educated decision rather
than just rush into the radiation. After looking at all the options my brother
chose not to proceed with the standard treatment of radiation and/or chemo.
Instead we began looking into alternatives including supplements, dietary
changes and Chinese medicine. We don't know the future, but the tumor is
stable eight months after diagnosis. We hope he will beat the odds, having
done something different.

111
Chapter 8: Radiosensitizers for DIPG
Chapter 8
Radiosensitizers for
DIPG
Roger J. Packer, MD
Radiation therapy remains the only eective treatment for brainstem gliomas
in children. Because its eectiveness is transient for most patients, attempts to
improve the benets of radiation therapy have been a focus of clinical research.
ese eorts have included increases in the total dose of radiotherapy delivered,
and alterations in the fractionation of radiation received (i.e., times per day
radiation is given and the dose at each delivery). ese modications to date have
not resulted in improved survival. e use of multiple small doses of radiation
per day to allow for a higher total daily dose (called hyperfractionated radiation
therapy) resulted in increased toxicity. Radiation damage to the brainstem (called
radionecrosis) is associated with increased neurologic decits.
Radiosensitizers
Radiosensitization is another means to improve the therapeutic balance between
ecacy and toxicity of radiation therapy. is research has been explored over
the past two decades, and continues to be studied. Radiosensitizers are dened as
compounds that, when combined with radiation, achieve greater tumor inactivation
than would have been expected from just the additive eects of the two modalities
of treatment. e premise that underlies radiosensitization is that the toxicities of
the chemical agent used and the radiation do not signicantly overlap, thereby not
increasing toxicity. Also, the chemical agent
chosen should not make the radiation therapy
more toxic to the normal brain cells within
the region of the brain receiving the radiation.
Optimally, the benets of radiosensitization
should allow the tumor site to be exposed to
an eectively higher dose of radiation without
increased toxicity. In reality, this hoped-for
Dr. Packer is the Director
of the Brain Tumor Institute
and Senior Vice-President of
Neuroscience and Behavioral
Medicine at Children’s National
Medical Center, and Professor
of Neurology and Pediatrics
at The George Washington
University, Washington, DC.
Chapter 8: Radiosensitizers for DIPG
112
113
Chapter 8: Radiosensitizers for DIPG
synergistic eect of radiosensitization is a balance between how much more eective
the radiosensitizers make radiation in killing more tumor cells, compared with how
much more damaging the treatment will be to normal cells that are exposed to the
radiotherapy.
Another basic concept that has been dicult to prove with brainstem gliomas is that
increasing the eective dose of radiation therapy actually improves disease control.
At best, most radiosensitizers increase the dose intensity of radiotherapy by 20%
to 30%, yet it is unclear whether such an increase results in improved long-term
disease control.
Types of Radiosensitizers
Hypoxic cell sensitizers
e largest early experience with radiosensitizers was the use of agents that
act to sensitize hypoxic cells to radiation therapy. e use of these agents was
based on the assumption that a chronic state of tumor hypoxia (when tumor
cells exist in an environment low in oxygen) occurs in brainstem gliomas.
Using agents that make these hypoxic tumor cells more sensitive to radiation
therapy may provide a therapeutic advantage by killing more hypoxic cells and
sparing better oxygenated cells (in theory, normal cells) from the damaging
eects of radiation. A series of hypoxic cell sensitizers have been utilized
for other cancers, but they have not been used in brainstem gliomas due to
concerns about toxicity, including enhanced neurologic toxicity, and lack of
any clear benet when used on other types of tumors.
An alternative means to radiosensitize hypoxic cells is to make the tumor less
hypoxic—in other words, get more oxygen to the cells. Dierent methods
that have been used to accomplish this include hyperbaric oxygen, the use
of red blood cell transfusions, and the delivery of oxygen carrier substances.
e combination of nicotinamide and carbogen has been used based on the
theory that nicotinamide will decrease the presence of intermittent hypoxia
(a deciency in the amount of oxygen reaching tissues), and the carbogen will
re-oxygenate tumor cells. Other approaches have included using drugs such
as nitric oxide, which causes vasodilation (widening of the blood vessels), to
alter the tumor vasculature.
Non-hypoxic cell sensitizers
Some of the earliest work in non-hypoxic cell sensitizers was the use of cell-
cycle-specic radiosensitizers that act independently of the eect of oxygen.
Drugs such as BUDR (bromodeoxyuridine) and IUDR (idoxuridine), which
are halogenated pyrimidine analogs, have been tested. It is presumed they
would primarily sensitize rapidly proliferating cells, with the drug sensitizing
tumor cells to a much greater degree than the normal surrounding cells. For
ecacy, these drugs require extended exposure to allow incorporation into
the tumor cell’s DNA. e use of halogenated pyrimidine analogs has not
been shown to be eective in adults with high-grade gliomas, and has not
been extensively studied in children.
Gadolinium-texafyrin is a molecule that penetrates well into enhancing
regions of the brain. Gadolinium is the primary contrast agent used for
magnetic resonance imaging (MRI) studies. Gadolinium-texafyrin is an
oxygen-independent radiosensitizer with a low toxicity prole. A phase I
dose-limiting toxicity study (see chapter 5) in children with brainstem gliomas
demonstrated minimal side eects. A phase 2 study of this drug regimen given
on a Monday through Friday schedule has recently been completed through
the Children’s Oncology Group (COG). Arsenic trioxide is another non-
hypoxic radiosensitizer which remains in phase 1 study for pediatric gliomas.
Chemoradiation
Chemotherapeutic agents have been widely used in children with brainstem
gliomas during radiation therapy in an attempt to radiosensitize the tumor
cells. e major diculties in choosing the most appropriate chemotherapeutic
agent to study include:
• e independent eectiveness of any drug in children with recurrent or
newly diagnosed brainstem glioma has been dicult to prove.
• Most phase 2 or pre-radiotherapy neoadjuvant studies (administration
of therapeutic agents prior to the main treatment) have demonstrated
minimal ecacy. Experimental data to support the synergistic benets of
chemotherapy, when added to radiation therapy in experimental models,
has been limited.
• e drug(s) may have limited ability to get to the tumor site because of
a relatively intact blood-brain barrier.
• e toxicity prole of the drug has to be “reasonable,” given the impaired
neurologic status and probable shortened life-span of the child, so as not
to detract from the remaining quality time that the child and family can
share.
Chapter 8: Radiosensitizers for DIPG
114
115
Chapter 8: Radiosensitizers for DIPG
Despite these considerations, there are multiple reasons to consider the use
of chemotherapeutic agents. For example, the radiation eect on tumor cells
may be enhanced by drugs that inhibit DNA repair after sublethal radiation
damage. Drugs such as cisplatin, hydroxyurea, and nitrosoureas have shown
this type of activity in experimental studies. Several drugs have been shown to
potentially enhance the ecacy of radiotherapy by changing the cell cycling
of tumor cells—putting more cells into a phase of the mitotic cycle, which
makes them more sensitive to radiation therapy.
Another potential benet of chemotherapy would be the independent eect
of the chemotherapeutic agent to decrease the size of the tumor cell. is
would potentially make radiotherapy more eective. Similarly, if radiotherapy
made the tumor smaller, it might allow greater vascular access for the
chemotherapeutic agent, resulting in a greater concentration of the drug in the
tumor and increased tumor kill. Some chemotherapeutic agents are relatively
eective in killing hypoxic cells. For this reason, drugs such as cisplatin may
have synergistic ecacy if used with radiation therapy. Chemotherapeutic
agents may also enhance apoptosis (programmed cell death) and subsequently
tumor death, especially in cancer cells sublethally damaged by concurrent
radiotherapy.
Delivery of Chemotherapy and Radiation
e timing of chemotherapy and radiation delivery is often based not only on
the principles of when the chemotherapy may be most eective in enhancing
radiation, but also the practicality of delivering such treatments to children. e
toxicity of some chemotherapy agents precludes treatment throughout the entire
6 to 7 weeks of prescribed radiation therapy. In addition, the need to transport
the child to the radiation therapy facility, which is often at an institution other
than where the chemotherapy has been given; the time needed to sedate young
children; and the concern of overburdening both the child and the family for
an unproven treatment, often results in severe logistical issues and compromises
in treatment schedules. In reality, these issues and compromises have resulted
in chemotherapeutic agents being given with radiation therapy in a sequential
fashion in order to reduce the temporal separation as much as possible.
e benets of chemoradiation have been suggested in a variety of dierent
tumor types, including head and neck tumors, small cell lung cancer,
gastrointestinal cancers, and cancers of the genital and urinary organs. However,
a clear-cut benet in adults with primary central nervous system tumors has
been dicult to prove. Pediatric tumor studies to date have been disappointing
as well, with the possible exception of medulloblastoma, where a preliminary
trial suggested improved survival.
Platinum derivatives
e use of platinum derivatives such as chemoradiation sensitizers is based
on the ability of drugs such as cisplatin and carboplatin to inhibit DNA
repair or sublethal radiation damage and to have cytotoxic eects on hypoxic
cells. Cisplatin has been used as a backbone of a study comparing standard
fractionated radiation to hyperfractionated radiation, without a clear survival
advantage in either arm of the trial. Another study utilized carboplatin twice
weekly during hyperfractionated radiation therapy without any clear-cut
benet. A subsequent carboplatin study coupled carboplatin with a bradykinin
derivative to enhance delivery of the carboplatin to the brainstem. In this trial,
carboplatin was given on Monday through Friday basis throughout radiation
therapy with good tolerability, but overall, survival did not dramatically dier
from that seen in historical controls treated with radiotherapy alone.
5-FU derivatives
e drug 5-FU has been extensively utilized in the treatment of adult cancers for
both its antineoplastic eects and radiosensitization properties. 5-FU interacts
with radiotherapy through disruption of cell kinetics and direct eects on
repopulation of cells. Experimental work has shown that 5-FU is truly synergistic
with radiotherapy. Its toxicity prole however, makes its concurrent use in
pediatrics dicult. A sister drug, Capecitabine, has recently completed phase
1 and phase II testing as a radiosensitizer given concurrently with radiation
therapy on a daily basis in children with brainstem gliomas. Results from the
data is pending.
Topoisomerase inhibitors
Etoposide is an oral topoisomerase inhibitor that has shown ecacy in one
trial of children with recurrent brainstem gliomas. e drug penetrates into
the central nervous system well. However, a COG phase II trial of etoposide
in combination with vincristine, concurrently with radiation therapy showed
no clear-cut benet.
Another topoisomerase inhibitor, topotecan, has been studied in a phase 1 study
concurrent with radiation therapy for children with newly-diagnosed brainstem
gliomas. A phase 2 study was initiated through the Children’s Oncology Group
but not completed.
Chapter 8: Radiosensitizers for DIPG
116
117
Chapter 8: Radiosensitizers for DIPG
Temozolomide
Results of adult trials with high-grade gliomas led to enthusiasm for the
use of temozolomide, concurrent with radiation therapy, for patients with
newly diagnosed brainstem gliomas. In children with recurrent brainstem
gliomas, temozolomide has not shown signicant independent activity and
there is little experimental data to show that temozolomide is synergistic with
radiotherapy. But based on the adult data and the relatively good toxicity
prole of temozolomide when used at a low dose on a daily basis, a study was
performed through the Children’s Oncology Group that coupled temozolomide
with radiation therapy, and followed the completion of radiotherapy. e results
of this study have been disappointing.
Radiosensitization with Molecular-Targeted Drugs and Other
"Biologic Agents"
Over the past decade, a host of biologic agents have become available and tested
in children with brain tumors. Molecularly targeted agents have included:
1. Antiangiogenesis drugs;
2. Agents that block growth factor receptors;
3. Drugs that interfere with intracellular signaling essential for tumor growth.
e lack of biologic information about brainstem gliomas has hindered, to a
great degree, a biologic rationale for deciding which drug, or drug combinations
would be most eective if combined with radiation. In phase 1 trials to date,
these biologic agents have demonstrated minimal ability to shrink tumors,
although some have resulted in possible prolonged stable disease. Because of
the relative low toxicity of many of these agents and, in some cases, theoretic
evidence that they may be at least additive—if not synergistic—with concurrent
radiotherapy, multiple studies have utilized biologics with, and following,
radiation therapy. ere is great interest in continuing such approaches, with
the caveat that experimental evidence for independent ecacy or synergy is
often minimal, at best.
alidomide and Interferon
alidomide is an agent that has been in clinical use for many years for a
variety of indications, including sedation and leprosy, and was found to be a
potent teratogen in pregnant women. Among its multiple properties, including
being an anti-inammatory agent, it is also an angiogenesis inhibitor, which
probably underlies much of its teratogenicity. It has been used in combination
with radiotherapy for children with brainstem gliomas without clear benet.
Another drug that was utilized with radiotherapy is interferon. Interferon,
of which various types including alpha, beta, and gamma being utilized, has
shown variable benets for adults and children with malignant gliomas. Based
on radiographic objective responses in 4 out of 18 children with recurrent
malignant cerebral or brainstem glioma, and clinical improvement or disease
stabilization in 5 out of 9 children with recurrent brainstem gliomas treated
on a beta-interferon protocol for children with recurrent disease, a study of 32
children with diuse intrinsic brainstem gliomas were treated with concurrent
recombinant beta interferon and hyperfractionated radiation therapy (7200
cGy). is study was disappointing in that not only did 30 of 32 patients
develop progressive disease at a median of 5 months from diagnosis, but more
than one-third of the patients required dose modications due to a hepatic
(liver related) or hematologic (blood related) toxicity. One patient developed
severe neurotoxicity.
Molecularly targeted trials
A variety of biologic agents have been studied concurrently with radiotherapy.
Iressa, an epidermal growth factor receptor antagonist, was studied in both
phase 1 and phase 2 studies through the Pediatric Brain Tumor Consortium
(PBTC). Early experience with this drug raised the issue of whether the use
of such agents would increase symptomatic brainstem hemorrhage. is study
highlighted how little was actually known about the rate of occurrence of
spontaneous hemorrhages of brainstem gliomas prior to, or during, conventional
radiotherapy. Intrabrainstem hemorrhages did occur, but it is unclear how much
more frequent their occurrence was than the rate of such episodes during and
after standard radiation therapy. Although there was no clear-cut benet from
the use of the drug, survival rates one year from study were more than 50%.
e farnesyl transferase inhibitors are drugs that interfere with intracellular
signaling by blocking the activation of ras—a key protein involved in
intracellular signaling often overactive in growing tumors. An oral farnesyl-
transferase inhibitor drug was utilized in phase 1 and phase 2 studies through the
Pediatric Brain Tumor Consortium with a low rate of intratumoral hemorrhage
and toxicity, but no evidence of ecacy. Antiangiogenic agents are likewise in
clinical use for patients with newly diagnosed brainstem gliomas. ere is little
data, as of yet, concerning their toxicity or ecacy.
Chapter 8: Radiosensitizers for DIPG
118
119
Chapter 8: Radiosensitizers for DIPG
Summary
Radiosensitization remains an appealing approach for the treatment of
children with newly diagnosed brainstem gliomas. As outlined, although there
are theoretic benets to their use, there is little evidence to date to validate
improved ecacy. Work continues with standard chemotherapeutic agents,
hypoxic and non-hypoxic radiosensitizers, and biologic agents. Until more is
known about the basic biology of brainstem gliomas, most studies will remain
empiric. Delivery of agents to the brainstem remains an extremely critical and
potentially limiting factor. In addition, as better agents are developed, their
selective capabilities of killing tumor cells and relatively sparing the normal
surrounding brain cells will remain a critical issue.
Parent Perspectives
When our daughter Ella was diagnosed with DIPG the only trial available
was a radiosensitizer. So we opted to do it because we had no other options.
Her first treatment was a couple weeks after diagnosis. She was to have
the radiosensitizer and then radiation within a couple hours. Within 30
seconds of the radiosensitizer entering her body she began sweating, having
stomach cramps and nausea. We asked the doctors to stop the treatment
and were told they would give her additional anti-nausea meds and anti-
anxiety meds. She finished the treatment, but due to the constant vomiting
she was unable to have radiation as she would be strapped to the table
and the fear was that she could choke on her own vomit. We opted out of
the trial the next day.
Although our journey began a day prior to April 11th, I mark that as the day
the world effectively ended for us. On that day, we were brought into a small
cramped room next to the nursing station, given no more than ten minutes
with the attending oncologist and told that Alexis, our then twenty-seven
month old daughter, had only six to nine months to live, maybe a year at
the outside. Wind taken out of our sails, devastated beyond belief, we were
effectively set out to drift in the new and confusing world of pediatric cancer.
We were barely familiar with the words diffuse intrinsic pontine glioma.
Within several hours, we found ourselves frantically driving from one
hospital to the next to meet with another doctor to discuss treatment options
and prognosis. Within a very short amount of time, we were provided with an
amount of hope that, although not a guarantee of survival, at least allowed
us to gain some level of comfort with moving forward.
During that initial meeting late on a Friday afternoon, we discussed several
approaches and treatments. Each option was presented with its own caveat,
and each was discussed in a manner which allowed us to appreciate that
it was not a home run cure. Ultimately, our decision was between one of
two treatment paths: a chemotherapy cocktail with overly used agents, or
standard radiation with a radiosensitizer. Ordinarily, chemotherapy has
proven ineffective for children with DIPG. The thought process behind this
option in Alexis’ case was based upon her age at diagnosis and a hunch
Chapter 8: Radiosensitizers for DIPG
120
121
Chapter 8: Radiosensitizers for DIPG
that her tumor was low grade. This option necessitated lengthy stays in the
hospital, which worried us beyond belief. Personally, we were concerned
about Alexis’ psychological welfare during the course of extended stays in
the hospital as well as her overall quality of life. In addition, this course
had no statistical efficacy. Truth is, we would have done anything in the
world to save Alexis, regardless of course.
Ultimately, the decision was made to proceed with a course of thirty
radiation treatments and a Phase II clinical trial utilizing a radiosensitizer.
The hope was that the addition of the radiosensitizer would allow the
radiation to be more effective. The theory behind radiosensitizers is that
the compounds are taken up by the blood vessels in the tumor and thus
direct the radiation directly to the tumor. At the time we chose this course of
treatment, there was no data presented to us, either statistical or anecdotal
in nature, and thus, it was simply a blind dart thrown at a wall. You hope
and you pray that the dart sticks. Hindsight taught us that in rare instances
a single dart would find its target with no rhyme or reason. Since Alexis
was very young at the time of diagnosis, we did not believe that she could
aide in any reasonable fashion with making critical decisions. Accordingly,
the guiding factor in all decisions was based upon Alexis’ quality of life.
At diagnosis, Alexis’ main symptom was an inverted right eye that was
infrequent in nature. While watching television, we noticed that Alexis had
to turn her head to the side to view. We feared that radiotherapy would cause
swelling and thus we would witness an increase in symptoms. Within the
first three days, these fears were realized and we noticed some right-sided
weakness. Obviously, this was extremely frightening and disconcerting.
Thankfully, this abated within a day or so of additional treatment and Alexis
sailed with flying colors through the remaining twenty six or so treatments
until she “graduated” from radiation on June 19th.
Our days during radiation therapy grew to be routine, and again, we
normalized life as much as possible. Each morning, we frantically rushed
to the hospital by 5:45 a.m. If we were late, the entire schedule and timing
of the actual radiotherapy was delayed. Upon arrival, we marched straight
to one of two treatment rooms on the pediatric hematology/oncology ward.
Shortly thereafter, Alexis received several pre-medications and then the
radiosensitizer was brought into the room and pumped into Alexis’ veins
over the course of fifteen minutes. The first several times she was given
this combination we sat with breath held, worried about what side effects
may present. Thankfully, none ever manifested. We of course were forced
to wear rubber gloves when changing Alexis due to the potential toxicities
to those who came in contact with the drugs. Such an odd juxtaposition—
Alexis could have these substances dripped into her veins, but there was
concern over us touching them with our bare hands. After the combination
was finished, we then waited three hours for the sensitizer to “find” its way
throughout her diminutive little body and on to the tumor. It was always
a tormenting thought: just inches inside of Alexis’ head sat this uninvited
beast. And nothing that we could do could ever change that. This realization
continually taunted me during my waking moments and beyond.
Alexis’ course was not typical in the DIPG community. Within several days
of beginning radiation in combination with the radiosensitizer, all symptoms
were erased and that remained true up until the end of January.
Our son was 10 years old when he was diagnosed with DIPG. Like most
10 year old boys, he was active and vibrant—full of life. He lived life to
the fullest and was happiest when he was playing baseball.
There weren’t many clinical trials available for children newly diagnosed
with DIPG. The only trial available via the pediatric oncology clinic easily
accessible for us was a Phase II COG trial involving motexafin gadolinium
as a radiosensitizer. We were told that radiation therapy is the only treatment
which usually makes a difference for kids with DIPG. Therefore, the most
logical focus in clinical trials was to increase the effectiveness of radiation.
This made sense to us, so we entered Caleb in the trial.
Each morning, Caleb received an infusion of the bright green motexafin
gadolinium compound. The hour-long drive to the clinic, the hour-long
infusion, and the two to five hour wait time before Caleb could have
radiation all turned out to be blessings to our family. We had good times
together during those days—time focused on Caleb and crafts, games or
reading. Often, Caleb’s brothers or his best friends would come along with
us. We developed dear friendships and learned to treasure the moments.
Within a week of beginning the trial, Caleb began to develop unusual side
effects. He became very sensitive to the sun; he felt a prickly sensation
that quickly developed into full-fledged pain when he was outdoors. Within
days of this, blisters began appearing on his face, hands, neck, arms, and
ears. At first they were just little water blisters. Soon, they became huge
bubbles—some of them as large as a small apple on the backs of his hands.
Chapter 8: Radiosensitizers for DIPG
122
His fingernails turned a milky white and began to separate from his nail
beds.
All the while, Caleb continued to play baseball. We saturated him in sun
screen and covered his skin with cloth and bandages. The pain was intense.
He could hardly stand to be in the field. After the third out was called, he
raced into the dugout where we had ice chests filled with cold and warm
rags in which to wrap him—one of the few efforts that brought some relief.
We rubbed topical anesthetic on his skin and, when it was time for him to
hit, he wore thick gloves to dull the pain of the blisters as his hands tightly
gripped the bat. He wore an eye-patch on his left eye. The green chemicals
gave his skin a green tint and even produced green stripes down his neck
and back and across his shoulders. Add the blisters, various bandages and
big gloves and he looked like quite a character. He was devoted to the game.
I greatly regret that we allowed him to suffer so—even required it of
him. He never complained about the therapy; never once did he consider
withdrawing from the trial. We believed it held promise and was the only
hope we had of beating the evil disease.
That does bring to mind regret, however. While he never considered stopping
the therapy, he did consider taking a year off from baseball. Now, I know
a lot of boys love baseball, but Caleb suggesting he take a year off from
baseball is akin to most of us taking a year off from food. It proves how
difficult it was for him. And, because we knew that he likely wouldn’t have
another year, we felt we had to explain to him that he had to make the most
of the current season. We were blunt—it might be his last season.
Again, in retrospect, I wish we hadn’t been so forthright. He increased his
resolve and will to fight. But it also caused him to be fearful at times. Caleb
surely would have eventually figured out his prognosis. I wish, though, that
the circumstances had been different—that we hadn’t forced the knowledge
on him out of desperation and baseless hope.
We do not blame ourselves or our healthcare team for these painful
memories. We do, however, wish that our healthcare team had been more
forthright with us and told us that radiosensitizers in general had never
shown much promise in DIPG, and that this specific therapy hadn’t helped
a single kid. It was just the only thing they knew to try at the time.
Who knows? We might have forged on, convinced that Caleb would be “the
one.” That’s what we all hope, isn’t it? But at least we would’ve known.

123
Chapter 9: Chemotherapy and Biologics
Chapter 9
Chemotherapy and
Biologics
David N. Korones, MD
e cure rate for children with cancer has improved dramatically over the past
few decades, rising from less than 50% in the 1960s to close to 80% today.
is is a remarkable story of laboratory research, which has given us a better
understanding of how children’s cancers work. It is the story of unprecedented
collaboration between hundreds of pediatric cancer centers around the country
and the world. It is the story of pediatric oncologists, radiation oncologists, and
surgeons working together to improve the lives of children with cancer. And it
is the story of strength, heartbreak, resilience, inspiration, and advocacy on the
part of children with cancer and their families, who have contributed more than
anyone in moving this eld of medicine forward.
Yet the successes in curing children with cancer have been unequal. Remarkable
strides have been made in treating some types of childhood cancers, but cure rates
for others remain stagnant. Such is the case for children with brainstem glioma, for
whom success has sadly eluded us. Surgical removal of this tumor is not possible.
In fact, these children rarely undergo a biopsy, so tumor samples are not available
to help researchers understand this disease. Radiation has proven to be quite
eective, but only for a short time. is leaves chemotherapy and biologics. It
would seem that for a tumor that cannot be removed surgically and that responds
only temporarily to radiation treatment, chemotherapy might be the best approach
to this challenging disease. But this has not
been the case. Despite decades of research
and clinical trials, researchers have yet to
nd a chemotherapeutic or biologic agent
that improves the survival of children with
brainstem glioma. But this does not mean
we never will. In fact, it could be a new
drug or some sort of targeted therapy based
Dr. Korones is Board Certied in
Pediatrics, Pediatric Hematology/
Oncology, and Hospice and
Palliative Medicine. He is Director
of the Brain Tumor Program
and the Pediatric Palliative Care
Program at Gosliano Children’s
Hospital, Rochester, NY.
Chapter 9: Chemotherapy and Biologics
124
125
Chapter 9: Chemotherapy and Biologics
on a better understanding of the biology of brainstem gliomas that will lead to
future success in treating children with this disease. at day has not yet come,
but it will. is chapter reviews what chemotherapy is, why it has not worked for
children with brainstem glioma, what has been tried, and what holds promise.
Also discussed are biologic therapies (i.e., treatment that more specically targets
some of the mechanisms tumor cells use to grow).
Chemotherapy
What is chemotherapy?
Chemotherapy is simply any medication that kills cancer cells. Just as
“antibiotic” refers to the broad class of drugs used to treat infections,
chemotherapy refers to the broad class of drugs used to treat cancer.
Chemotherapy can be given intravenously (IV), through an injection into
the skin or muscle, orally, or even injected directly into a body cavity (e.g.,
injection of chemotherapy into the spinal uid via a spinal tap [lumbar
puncture]). Most chemotherapy is given by IV or orally. Chemotherapy works
by targeting cells that are actively dividing thereby stopping the cancer cells
from reproducing. Many dierent types of chemotherapy drugs exist, and
each one targets one of the many dierent aspects of tumor cell division. For
example, two commonly used chemotherapies to treat children with brain
tumors, temozolomide and CCNU (lomustine), bind directly to DNA (the
building block of cell division) and prevent the DNA from duplicating itself,
thereby preventing the cell from dividing, leading to its death. Another
example is vincristine, which targets spindle-like structures that allow one
cell to become two cells.
For many pediatric cancers, it has been shown that by using combination
chemotherapy—two, three, or more drugs together—the child’s survival is
improved. is is because several drugs together work better to kill cancer
cells than any one drug alone. In addition, if a tumor is resistant to one drug,
it may be killed by one of the others given.
A disadvantage of chemotherapy (particularly in treating children with brain
tumors) is that it circulates everywhere in the child’s body, and therefore has
the potential to kill or damage normally dividing cells. is fact accounts for
many of the side eects of chemotherapy, such as low blood counts, risk of
infection, bleeding, fatigue, mouth sores, and nausea and vomiting.
Challenges of using chemotherapy for children with brainstem glioma
Finding effective chemotherapy for children with brain tumors is more
challenging than nding eective chemotherapy for children with other
malignancies. A major challenge with brain tumors is the blood-brain barrier.
Blood vessels in the brain are unique; they are designed to be very selective
about what can penetrate them to get into brain cells. ey selectively allow
nutrients to reach brain cells, but block many unrecognizable, potentially
toxic substances including many types of chemotherapy. From an evolutionary
standpoint, this makes perfect sense (i.e., protecting our brains from toxins),
but when it comes to getting chemotherapy into brain tumors, it is a problem.
erefore chemotherapy must be designed to penetrate the blood-brain barrier.
e list of drugs that can do this is small, leaving us with fewer weapons to use
for children with brain tumors. It is thought (but not proven) that the blood
vessels in brainstem gliomas are particularly restrictive and allow very few
substances to penetrate them.
Another challenge is the tumor itself. For reasons that are unclear (and still not
denitely proven), it appears that the tumor cells that make up brainstem gliomas
are extremely resistant to chemotherapy. at is, even if the chemotherapy gets
into the tumor, it cannot kill the tumor cells.
Clinical Trials Using Chemotherapy for Children with
Brainstem Glioma
As noted above, chemotherapy has been used in every way, shape, and form
possible to date to treat children with brainstem glioma. e most common
approaches have been giving chemotherapy after radiation (termed adjuvant
chemotherapy), administering chemotherapy before radiation (called
neoadjuvant chemotherapy), and giving high-dose chemotherapy with stem cell
rescue (also referred to as autologous bone marrow transplant). Chemotherapy is
also used during radiation treatment as a radiosensitizer (discussed separately in
chapter 8). ese various approaches to using chemotherapy are discussed below.
Adjuvant chemotherapy
The first large-scale multicenter clinical trials testing the effectiveness of
chemotherapy for children with brainstem glioma were published in the late
1980s. is was around the time that chemotherapy had been proven eective
for children with other types of brain tumors, such as medulloblastoma,
glioblastoma and low-grade astrocytoma. At that time there was considerable

Chapter 9: Chemotherapy and Biologics
126
127
Chapter 9: Chemotherapy and Biologics
hope that the successes of chemotherapy for children with other brain tumors
would extend to children with brainstem glioma. In one of the rst studies,
Jenkins et al. conducted a national clinical trial through the Children’s Cancer
Study Group (CCSG) from 1977 to 1980. All 74 children enrolled in the study
received radiation; half received radiation alone, while the other half received
radiation plus chemotherapy—lomustine (CCNU), vincristine, and prednisone
(a combination used successfully for children with medulloblastoma and high-
grade astrocytoma). e 5-year survival for both groups was only around 20%,
suggesting that this particular combination of chemotherapy was not eective for
children with brainstem glioma. It should be noted that survival in both groups
was actually somewhat higher than what we see today. is is probably because
the study was conducted in the pre-MRI era, and some of the tumors that were
thought to be brainstem glioma may have actually been less aggressive variants
of brain stem tumors that would not be classied as DIPG today.
rough the 1990s, researchers explored other types of chemotherapy for
children with brainstem glioma. Walter et al. treated nine children with an MRI-
documented DIPGs with carboplatin and etoposide during and after radiation.
ese two drugs have been shown to have activity against other types of brain
tumors, so the hope was that they would also prove eective for children with a
brainstem glioma. However, this treatment did not work—survival at 1 year was
44%, and at 2 years only one child (11%) was alive. In another study, Chamberlain
tried an innovative approach to treatment, which comprised daily low doses of
oral chemotherapy. e thought was that daily low doses of chemotherapy given
orally might work better than larger IV doses of chemotherapy given every few
weeks. In addition, side eects seemed to be less of a problem when this type of
dosing had been used in other clinical trials. Chamberlain used this approach,
treating 12 children with a recurrent brainstem glioma with oral etoposide
(VP-16). Unexpectedly, the tumor shrunk in 5 of these 12 children. is result
generated considerable excitement about this new approach to treatment. Based
on Chamberlain’s observations, the Children’s Oncology Group (COG) launched
a national clinical trial of oral etoposide and IV vincristine during and after
radiation. Disappointingly, all 30 children on this trial died, with one and two
year survival of only 27% and 3% respectively. Other types of chemotherapy
have been tested, mostly in small trials, and they, too, were ineective. Drugs
including idarubicin, trophosphamide and oral etoposide, cisplatin, etoposide,
ifosfamide, and oral topotecan have been tried, but none prolonged children’s
lives or improved cure rates. A summary of these trials is illustrated in Table 1.
One exciting new chemotherapeutic agent developed and studied extensively in
Table 1: Representative clinical trials of adjuvant chemotherapy for children with newly diagnosed diuse brain stem
glioma.
Author (see
Appendix D)
Group study
N RT dose
(GY)
Chemotherapy Median
OS (mo.)
Survival (%) at
1 yr 2 yr 3 yr 5 yr
Jenkin (1) CCG 33
37
50-60
50-60
none
pCV
26* 17
35* 23
Walter (2) St. Jude 9 70.2 (1.17
b.i.d)
carboplatin
IV VP-16
10 44 11
Korones (4) POG 30 54 (1.8
q.d.)
oral VP-16
vincristine
9 27 3
Wol (6) GPOH 20 54 (1.8
q.d.)
oral trophosphamide
oral VP-16
8 40 15 5
Wol (7) GPOH 37 54 cisplatin
IV VP-16
Vincristine
13
Broniscer (9) St. Jude 33 55.8 temozolomide 12 48
Cohen (10) COG 63 temozolomide 10 40
* 18 mo. survival
CCG = Children’s Cancer Group POG = Pediatric Oncology Group COG = Children’s Oncology Group
GPOH = German Society of Pediatric Oncology RT = radiotherapy pCV = prednisone, CCNU, vincristine OS = overall survival
Chapter 9: Chemotherapy and Biologics
128
129
Chapter 9: Chemotherapy and Biologics
the last 10 to 15 years is temozolomide (Temodar®). is drug is given orally,
has fewer side eects than most chemotherapy, and has proven to be eective
for adults with grades 3 and 4 astrocytomas (similar to brainstem glioma). e
successes with adults led pediatric neuro-oncologists to conduct clinical trials
of this drug for children with brainstem glioma. In one of the rst studies of
temozolomide for children with a newly diagnosed brainstem glioma, 33 children
at St. Jude Children’s Research Hospital received temozolomide in monthly cycles
for up to 6 months. Although the 1-year survival was 48% (higher than in most
studies), all 33 children eventually died. e COG conducted a similar study
with children with brainstem glioma receiving temozolomide during radiation, as
well as following it. e results were similarly disappointing. Of the 63 children
enrolled, all but one had died after 25 months of follow-up (and that child could
not be tracked down to verify whether he/she survived). In ve other studies of
temozolomide for children with a DIPG, (including one using cis-retinoic acid and
another using thalidomide), the outcomes were also poor. In sum, as promising
as temozolomide rst appeared to be, and as few side eects as it has, it is not the
breakthrough in chemotherapy we had so hoped it would be.
Neoadjuvant chemotherapy
As noted above, neoadjuvant chemotherapy is chemotherapy that is given before
the more denitive treatment for a tumor. In the case of a brainstem glioma,
neoadjuvant chemotherapy is chemotherapy given before radiation. is approach
was investigated in several clinical trials in the 1990s and early 2000s. e rationale
for neoadjuvant chemotherapy was based on the general enthusiasm at that time
for chemotherapy for children with brain tumors. In addition, it was felt that the
best way to assess the eectiveness of chemotherapy against a brainstem glioma was
to give it before radiation, as it is hard to assess tumor response to chemotherapy
after radiation. e hope was that eective drugs would quickly be identied and
then given to children after the radiation, as well. is approach was undertaken
with some trepidation, because if chemotherapy was not eective, the children
might suer from side eects of chemotherapy and a growing tumor, and get
sicker, not better. ere was concern that these children might become too sick
to tolerate radiation, the one treatment known to give them a little more time.
e rst clinical trial published taking this approach was a Pediatric Oncology
Group study of 32 children with brainstem glioma who were treated with two
to three cycles of cisplatin and cyclophosphamide before starting radiation.
Although 3 of the 32 children had shrinkage of the tumor and another 23 had
no increase in tumor size, the overall survival for the children was not improved
compared with giving radiation alone; the median survival was only 9 months. In
Table 2: Clinical trials of neoadjuvant chemotherapy for children with newly diagnosed diuse brain stem glioma
Author
(
see Appendix
D)
Group
study
N Chemotherapy Response RT dose
(Gy)
Median
OS (mo.)
Survival (%) at
1 yr
2 yr 3yr -
Kretschmar
(16)
POG 32 cisplatinum
cyclophosphamide
3 PR
23 SD
6 PD
66 (1.1 b.i.d.)
9 30
Jennings
(17)
CCG 32
31
carboplatin
VP-16
vincristine
cisplatinum
cyclophosphamide
VP-16
vincristine
2 PR
1 MR
12 SD
12 PD
1 PR
4 MR
8 SD
9PD
72 (1 b.i.d.)
72 (1 b.i.d.)
10*
10*
30* 18*
30* 10*
Doz (18) SFOP 36 carboplatin 4 MR
6 SD
11 PD
54 11
Broniscer (9)
St. Jude 16 irinotecan 10 SD
5 PD
55.8
*estimated from Kaplan-Meier curve PR=partial response, MR=minor response, SD=stable disease, PD=progressive disease

Chapter 9: Chemotherapy and Biologics
130
131
Chapter 9: Chemotherapy and Biologics
a second, larger study of 63 children with a newly diagnosed brainstem glioma, 32
children received carboplatin, etoposide, and vincristine, and 31 received cisplatin,
cyclophosphamide, etoposide, and vincristine before going on to receive radiation
therapy. Only 10 to 20% of the children had tumor shrinkage, and again, their
overall survival was poor—no better than it was with radiation alone. Similarly
poor results were seen when 38 children received neoadjuvant carboplatin and
neoadjuvant irinotecan. ese studies are summarized in Table 2.
Based on these disappointing results and the disappointing results of multiple
other clinical trials of chemotherapy after radiation, the approach of trying
chemotherapy before radiation has largely been abandoned.
High-dose chemotherapy and stem cell rescue
Another innovative approach undertaken in the quest to cure children with
brainstem glioma was high-dose chemotherapy with stem cell rescue (also referred
to as autologous bone marrow transplantation). is type of therapy is not really
a bone marrow transplant at all; rather, it is simply a way to more safely give
children higher doses of chemotherapy. e rationale for using such high doses
of chemotherapy for children with brainstem glioma is two-fold. It was thought
that 1) it might take higher doses of chemotherapy to kill the brainstem glioma
cells, and 2) higher doses might somehow “push” more chemotherapy through
the blood-brain barrier and allow enough chemotherapy to reach the tumor cells
and kill the tumor.
Neuro-oncologists were particularly hopeful about this approach because it
seemed to improve survival of children with recurrent medulloblastoma and
glioblastoma. How does it work? First, the very youngest of blood cells (called
hematopoietic stem cells) are removed (often referred to as “harvested”) from
children with brainstem glioma. is is done by taking samples of bone marrow
or removing these specialized cells from the blood through a procedure called
pheresis. ese cells are frozen until they are needed. e children then receive 3
to 7 days of extremely high doses of chemotherapy, so high in fact that it almost
destroys their ability to ever make their own blood cells again. But after they
receive the chemotherapy, the stem cells that were removed and frozen are thawed
and given back to the children. ese resourceful stem cells are given through an
IV (like a blood transfusion), circulate through the blood, nd their way back to
the bone marrow, and over the next several weeks, repopulate the bone marrow
and the blood with new, normal blood cells, thus “rescuing” the children from
what otherwise could be a lethal dose of chemotherapy.
Several clinical trials of high-dose chemotherapy with stem cell rescue were
Table 3: Clinical trials of high-dose chemotherapy with stem cell rescue for children with newly diagnosed diuse brain
stem glioma
Author (see
Appendix D)
Group
study
N RT dose
(Gy)
Chemotherapy Median
OS (mo.)
Survival (%) at
1 yr 2 yr 3 yr
Bouet (21) SFOP 35 50-55 busulfan
thiotepa
10 40
Bouet (22)* France 5 not stated BCNU 10
Jackaki (23) Indiana U 6 54-59.4 procarbazine
CCNU
vincristine
13
Kedar (24)* U Florida 6 75.6 (1.25
b.i.d.)
cyclophosphamide
thiotepa
12.5
Dunkel (25)* CCG 6 72-78 (b.i.d.) BCNU
thiotepa
VP-16
11.4
*Chemotherapy given prior to radiation
Chapter 9: Chemotherapy and Biologics
132
133
Chapter 9: Chemotherapy and Biologics
A number of clinical trials of biologic therapies have been conducted. One of
the rst trials tested interferon, a natural substance made by the human body
in response to viral infections. It was hoped that interferon would stimulate the
immune system to ght the tumors and perhaps inhibit the blood supply to
the tumor. In a 1991 phase I-II clinical trial that included eight children with
brainstem glioma, two children had partial responses to the treatment, and in
another three children, growth of the tumor was halted for several months. In
a subsequent larger study of 32 children with a DIPG who received interferon
along with radiation therapy, overall survival was not improved.
Another innovative approach is using agents that can “open up” the blood-
brain barrier. As previously noted, chemotherapy has diculty reaching brain
tumors because it cannot penetrate brain tumor blood vessels. A substance called
RMP7 was found to open up these blood vessels, making them “leaky,” so that
chemotherapy can go right through them and reach the tumor. ere were
two very small clinical trials for children with brainstem glioma using RMP7
along with chemotherapy. In one phase I study of RMP7 and carboplatin, the
treatment was well tolerated but the number of children was too small to assess
its eectiveness. In a second study, eight children with brainstem glioma received
RMP7 along with various types of chemotherapy through an artery (instead of
intravenously). ese children did somewhat better, but again the number treated
was small and the intra-arterial approach is not one that can be easily done at
most centers. RMP7 was also studied in adults with malignant brain tumors
and was not found to be eective. e drug has limited availability at this time.
One additional approach to penetrating the blood-brain barrier is the use of
cyclosporine A along with chemotherapy. is drug helps “trap” certain types
of chemotherapy inside brain tumor cells so the chemotherapy remains in the
cells long enough to kill them. A COG trial using cyclosporine, vincristine, and
oral etoposide (VP-16) was launched to study this eect, but unfortunately, the
number and severity of side eects was unacceptably high.
Another promising biologic approach is anti-angiogenic therapy. is is the use of
drugs that kill or inhibit blood vessel growth into growing tumors. e rationale
for anti-angiogenic therapy is that if one can prevent blood vessels from growing
into the tumor, the tumor will be deprived of oxygen and nutrients and stop
growing. e most promising anti-angiogenic agent to date is bevacizumab, an
antibody that attacks a protein called vascular endothelial growth factor (VEGF).
VEGF is a natural substance that stimulates blood vessels to grow into brain
tumors as the tumors themselves get bigger. is treatment has proved eective
in adults with high-grade astrocytomas, and there was hope it might also prove
conducted in the 1990’s, but unfortunately this approach did not seem to help.
Perhaps the largest clinical trial using this approach was a study by Dr. Bouet
and colleagues. e researchers enrolled 35 children in the study, all of whom
received radiation, and 24 of whom were able to then go on and receive high
doses of chemotherapy (busulfan and thiotepa). All 24 of these children died,
three from complications of the therapy, and the remainder from regrowth of the
tumor. e average life expectancy of the group was only 10 months, not any
better than the life expectancy of children who received radiation alone. Four other
studies of high-dose chemotherapy with stem cell rescue included children with
brainstem glioma. Although tumors shrank in a handful of children, it did not
improve their overall outcome, and several children died of complications from
this very aggressive approach. A summary of trials using high-dose chemotherapy
with stem cell rescue is presented in Table 3.
Based on the experience with these children, high-dose chemotherapy with stem
cell rescue is no longer being investigated for children with brainstem glioma.
Although high doses of chemotherapy can be very eective for children with
certain types of cancer, it is clearly not the case for children with brainstem glioma.
We need other innovative approaches to tame this type of tumor.
Biologics
As is painfully clear from the above discussion, chemotherapy does not appear
to be the answer for children with brainstem glioma. Over the past decade,
investigators have come to realize the answer may lie in rst gaining a better
understanding of how these tumors work. Once we have that understanding,
we can nd drugs that target the tumor cells very specically; this is the concept
of biologic therapy. Chemotherapy is a rather blunt sword that kills any cell
that divides, be it cancerous or not. Biologic therapy is more nely tuned and
targeted. It is treatment that is based on the abnormal biology of a cancer cell.
Biologic therapy targets the very biologic mechanisms that cause cancer cells to
grow uncontrollably, and unlike chemotherapy, biologic therapy often spares the
normal cells.
A big challenge in using this approach for children with brainstem glioma is that
we do not know what makes these tumors tick. ese children seldom undergo
biopsy of their tumors, so researchers do not have many tumor samples to examine
to help them understand the biology of brainstem glioma. However, this is starting
to change, as some centers are doing biopsies again, others gather tumor samples
at the time of autopsy, and new techniques are being used to analyze old tumor
samples obtained decades ago.
Chapter 9: Chemotherapy and Biologics
134
135
Chapter 9: Chemotherapy and Biologics
eective for children with brainstem glioma. In a recently published study of
bevacizumab and the chemotherapy drug CPT-11 (irinotecan) for children with
recurrent brainstem glioma, only 5 of 13 children had a temporary halt in the
growth of their tumors, and the average time until the tumor starting growing
again was only 2.3 months. Although this particular anti-angiogenic therapy had
little eect, there are many new anti-angiogenic drugs being developed, so there
is still hope that this approach may eventually work.
Another hopeful approach to treatment is blocking growth factor receptors on
tumor cells. Growth factor receptors are proteins found on the surface of tumor
cells. When these growth factor receptors bind with a matching protein called
a growth factor, the two proteins link together and stimulate tumor cells to
grow uncontrollably. Based on studies of old tumor samples from children with
brainstem glioma, investigators have learned that brainstem glioma cells seem to
have increased numbers of a growth factor receptor called epidermal growth factor
receptor (EGFr). Clinician-investigators are hopeful that EGFr may be a new way
to attack these very resistant tumors. For example, in Germany, an antibody to
the EGFr was developed; this antibody (called nimotuzumab) binds to the EGFr
and seems to prevent EGF from binding to it and stimulating tumor cell growth.
In a clinical trial of nimotuzumab, 22 children with a recurrent brainstem glioma
were treated; 9 had stable disease and one child’s tumor actually became smaller.
Based on these encouraging results, a larger study of this drug was conducted,
but the results are not yet available. In another recently published clinical trial,
43 children with newly diagnosed brainstem glioma were treated with erlotinib,
another inhibitor of EGFr function. is drug was well tolerated, and children
on this study fared slightly better than average, including three children who
are alive more than 3 years from diagnosis. Blockage or inhibition of the EGFr
remains a hopeful approach for treating children with brainstem glioma, but
much work still needs to be done.
Other biologic therapies have been tried, but with no major successes. Some
(but not all) of the agents studied include tamoxifen, cis-retinoic acid, and
antineoplastons. e COG and Pediatric Brain Tumor Consortium have taken the
lead in the United States in exploring new and innovative approaches to treating
children with brainstem glioma. e COG is currently studying the addition of
vorinostat, a drug representing a new class of biologics called histone deacetylase
inhibitors (they unravel DNA), to standard radiation therapy.
Looking to the Future
As noted in the above discussion, clinician-scientists have exhaustively investigated
many dierent combinations of chemotherapy and radiation for children with
brainstem glioma. Sadly, after all this, they have discovered more about what
does not work than what does. We have learned that radiation works for a short
time, but chemotherapy does not seem to help at all. We have also learned that
there is genuine hope on the horizon. We now have the tools to examine the most
intricate details of the inner workings of brain tumor cells, including those of
brainstem glioma. Furthermore, we have a rapidly growing arsenal of new biologic
therapies that may be able to target the abnormal molecules/DNA/proteins that
cause brainstem glioma cells to grow. It is dicult to comprehend how hard it
must be for a parent, grandparent, family member, or friend to thumb the pages
of this book and read through the long list of therapies that do not work. But
we are on the verge of developing a list of therapies that will work, and our hope
is that with the next edition of this book, family and friends will read through a
long list of all the treatments that do work.
Chapter 9: Chemotherapy and Biologics
136
137
Chapter 9: Chemotherapy and Biologics
Parent Perspectives
Treatment, well really at this point there is none. You are told that steroids
and radiation will most likely shrink the tumor initially and help with
symptom management. Then you will get your “honeymoon period” before
the tumor begins to grow again and how soon that will be is anybody’s guess.
Chemo you ask? Yes of course, try this, or this, or this, but we have little
to no data indicating that any are effective in extending the life expectancy
of the child beyond 24 months. And well there is quality of life to consider.
We also opted to do chemotherapy in the hopes that in combination there
might be some added value. We agreed to known chemotherapies that were
generally well tolerated by children. I'm not sure we can ever be certain
how well they helped Liam's overall condition however, he had few side
effects. He took all his pills orally like a champ.
When scans showed that Liam's tumor had progressed on the combination
chemotherapies, we tried another course of a different combination therapy.
However Liam developed an allergic reaction to one of the medications and
we had to stop. His latest scan showed even further progression in parts of
his brain far from the confines of the brainstem. We tried one more drug and
it would prove to be his final chemotherapy. By this time Liam was not able
to walk at all on his own. He had significant hearing loss, and his speech,
when he spoke at all, was very hard to understand. He was growing tired.
When it was discovered that this treatment too did not have the effect that
was desired we decided to end treatment and begin palliative care. It was
not an easy decision but one that needed to be made for Liam.
As a result of our son’s chemotherapy we saw slow, steady improvement
of symptoms for over six months. But we were faced with a new challenge
during this time, a complication of long term steroid usage. Our son’s skin
began to thin, and his stretch marks opened. We were horrified to realize that
the treatment regimen (Avastin) that had made such a dramatic difference
against the cancer was delaying the healing of the open stretch marks. In
time this issue became more of a problem than the cancer itself, and we had
to face the reality that losing our son to wounds would be no less difficult
than losing him to brain cancer.
Our daughter underwent 6 weeks of radiation and chemotherapy. She did
not start steroids until she was almost complete with her treatment. She had
finished her treatment and 4 weeks later had to have a shunt placed due to
increased pressure. The following week she had her follow-up MRI from
the treatment and they saw new tumor growth in another area of her brain.
That weekend we went back to the ER and she had a seizure overnight. She
went into a coma and only came out once long enough to open her eyes and
we were able to look into her beautiful eyes again and tell her how much
we love her. She passed away exactly 3 months and 12 days from diagnosis
even with a full treatment of radiation and chemotherapy.
We used Capecitabine, 1300 mg. daily for sixteen weeks. There were
absolutely no benefits, just side effects. Joseph had hand and foot syndrome,
gas, and diarrhea.
I knew that at that time there were no known chemo treatments that worked
and had terrible guilt using my son as an experiment. My wife needed to
be doing something to save our boy. I wish the doctors would have been
clearer about the odds of this chemo working. I would have stopped much
earlier and concentrated on quality of life over treatments.
It was exactly one year after diagnosis that we learned the tumor was
growing again. With Andrew’s only symptom being headaches, we chose
to attempt to slow the progression of the tumor with a chemo cocktail of
vincristine, irinotecan and temozolomide given every three weeks. After
two cycles the scan showed continued growth, and we made the decision
to try temozolomide at full dosage in 28 day cycles in a second attempt
to slow tumor growth. We celebrated Christmas at home with a bald (but
happy) Andrew.
A scan in January showed further progression. Andrew’s doctor wondered
if the combination of Avastin and irinotecan might have a positive impact
against the tumor. We forged ahead with the new treatment plan, but felt
that Andrew was slipping away from us. We were pleasantly surprised
Chapter 9: Chemotherapy and Biologics
138
when Andrew’s scan in March showed a significant response to therapy:
shrinkage of the tumor, resolution of much of the cystic component and less
enhancement. We had hoped for slowed progression or possible stability;
instead we received an unexpected gift of time. Having already made peace
with what we thought was Andrew’s imminent death we were faced with the
challenge of changing our mindset back to one of living rather than dying.
We decided not to do chemo because at the end of the day we didn't see
much benefit to it. We didn't want Peyton being sick for what would be left
of her short life. We didn't want her suffering more than needed. We just
felt that the side effects of chemo outweighed any benefit the drugs "may"
have provided.

139
Chapter 10: Use of Steroids in DIPG
Chapter 10
The Use of Steroids in
Patients with DIPG
Eric Bouffet, MD
Ute Bartels, MD
Steroids play a major role in the management of patients with diuse intrinsic
pontine glioma (DIPG), particularly at the time of presentation. Corticosteroids
have been used to control cerebral edema in various conditions, particularly in
the context of aggressive brain tumors. Dexamethasone is generally considered
the steroid of choice because of its superior brain penetration and longer half-
life (time it takes for a drug to lose half of its pharmacologic activity). e role
of steroids in the management of disease at the time of progression and during
palliative care remains controversial. e aim of this chapter is to review the role
of steroids during the care of patients with diused pontine glioma.
Steroids During the Early Management of DIPG
At the time of diagnosis, steroids are usually the rst treatment oered to patients
with diuse intrinsic pontine glioma. Although their role has never been properly
assessed, most physicians prescribe steroids, and in particular dexamethasone,
once the diagnosis of DIPG is established. e doses used may vary individually
according to the clinical signs and symptoms of the child, but many physicians use
large doses, up to 10 mg/m
2
/per day in two or three doses. e aim of the treatment
is to a) improve neurological symptoms, b) reduce the edema surrounding the
tumor that may sometimes impact the ow of the cerebral spinal uid (CSF) and
cause some degree of hydrocephalus, and
c) prevent or minimize the edema induced
by the initiation of the radiation treatment.
Traditionally, physicians would keep the dose
of dexamethasone unchanged during the rst
week of treatment, and would then gradually
decrease the dose as neurological symptoms
Dr. Bouffet is the Director of the
Neuro-oncology Program and
a Senior Associate Scientist in
the Research Institute at the
Hospital for Sick Children, as well
as Professor of Pediatrics at the
University of Toronto, Canada.

Chapter 10: Use of Steroids in DIPG
140
141
Chapter 10: Use of Steroids in DIPG
improve with the radiation treatment. Some physicians prefer to keep a high dose
of steroids maintained throughout the 6 weeks of radiation therapy.
Steroids, however, are associated with signicant side eects that may aect the quality
of life of patients with diused pontine glioma. One of the most signicant side eects
is hyperphagia—a feeling of extreme excessive hunger. As a result, children with DIPG
often experience signicant weight gain during the rst weeks of treatment. e use
of steroids is also associated with personality changes, such as mood swings, anxiety or
sometime aggressiveness (see below). Cutaneous complications are not uncommon,
particularly in teenagers who can develop severe acne and stretch marks (or striae).
All of these side eects will improve within a few weeks of decreased or discontinued
use of steroids. ey will persist if the steroids are continued throughout treatment.
Steroids Following Radiation
Up to 50 percent of DIPG patients will present with symptoms of so called
“somnolence” in the weeks following completion of radiation treatment. e
onset of these symptoms is usually observed 2 to 4 weeks after the last session
of radiation, but can occur earlier or later.
e somnolence syndrome consists of symptoms ranging from mild drowsiness
to marked lethargy with prolonged periods of sleep, irritability, anorexia, low
grade fever, nausea and vomiting, cerebellar ataxia, dysarthria, dysphagia, and
headaches. For many parents who have not been informed of this complication,
the somnolence syndrome is suggestive of the initial manifestations of the
disease. e physiopathological mechanisms of this complication are not fully
understood, but somnolence is thought to be related to radiation-induced
disruption of myelination. e period of somnolence usually lasts 2 weeks,
and symptoms usually subside spontaneously. However, some patients can
experience symptoms for up to 4 to 6 weeks. When symptoms are signicant,
the use of steroids can be benecial. e optimal dose of steroids needed in this
context is unknown. However, spectacular improvement of both appetite and
sleepiness can be observed with small doses of dexamethasone, in the range of
0.5 to 1 mg per day.
Steroids at the Time of Progression
Because of their benecial eect at the time
of initial diagnosis, steroids are often used
at the time of recurrence of symptoms and
during palliation of DIPG patients. ey
often provide a marked improvement of recurrent neurological symptoms and are
usually prescribed with the short objective to relieve the symptoms of progression.
eir ecacy, however, is generally transient, and progressive symptoms recur
within one or two weeks following the prescription of the corticosteroids. At
this stage, the dilemma is whether to further increase the dose to alleviate these
symptoms or discontinue the steroids because of their potential adverse eects.
e choice is not easy for physicians and families and the decision should be
made with a clear understanding of the consequences of prolonged use of steroids
during palliation.
Although they can provide a transient improvement of neurological symptoms,
steroids have side eects that are of particular concern in the context of progressive
DIPG. Increased appetite and the resulting hyperphagia can lead to massive
weight gain and body transformation, in particular the cushingoid appearance
with the classic moon face. ese changes can have signicant cosmetic and
social implications leading to stigma and isolation. ey can also aect parents,
siblings and relatives at the time of bereavement when they remember the
cosmetic consequences of steroid usage. In addition, specic aspects of palliative
care and symptom progression of DIPG patients need to be taken into account
when steroids are considered. Lower cranial nerve decits lead to swallowing
disturbances and a risk of choking (see chapter 3). In this context, steroids can
have an unwanted eect, as they increase the appetite and therefore the risk of
choking of a permanently hungry patient. As swallowing disorders worsen, the
eects of steroids on appetite can become a nuisance and an obsession for the
child who is unable to eat or drink. In addition the combination of oro-pharyngeal
stasis of secretions and immunosuppression often leads to the development of
painful thrush that will require specic management with mouth wash and in
some cases antimicrobial medications against Candida.
Other side eects of steroids can have a signicant impact on the quality of
palliative care. In particular the behavioural side eects can alter the quality of
the interaction of the patient with his family.
Overall, the decision to use steroids at the time of progression should be made
with a clear understanding of the potential consequences of this choice. is
includes the risk of facing a vicious cycle as the progressive deterioration of the
disease leads to increasing the dose of steroids and subsequently to increasing the
side eects. Management of DIPG patients without steroids is possible, and some
neuro-oncology teams prefer to avoid their use at the time of disease progression.
Dr. Bartels is an Oncologist in
the Neuro-oncology Program at
the Hospital for Sick Children,
and Assistant Professor at the
University of Toronto, Canada.

Chapter 10: Use of Steroids in DIPG
142
143
Chapter 10: Use of Steroids in DIPG
Side Eects of Corticosteroids
Steroids are associated with a number of potentially serious side eects. e
onset and severity of these side eects usually correlate with the dose and
duration of the treatment. In the context of a short duration of administration
(2 to 3 weeks), most side eects will resolve after cessation of corticosteroid use.
However, some children may require prolonged steroid treatment because of
persistent or recurrent symptoms, or some physicians are reluctant to decrease
steroids during radiation. In this context side eects may persist or even worsen
over time and signicantly aect the child’s quality of life. Using the lowest
possible dose of steroids will reduce the risk of these complications.
Cosmetic side eects include cushingoid appearance, truncal obesity, hirsutism
(excessive hair), acne, and stretch marks. Other side eects include increased
appetite, immunosuppression, hypertension, glucose intolerance, electrolyte
disturbance, uid retention, peripheral edema, gastrointestinal side eects,
osteoporosis, avascular necrosis, growth retardation and ocular problems.
Complications of corticosteroids are summarized in Table 1. Among the
common side eects of steroids, weight gain, steroid myopathy, Pneumocystis
carinii pneumonia (PCP) and behavioral changes are of particular concern in
DIPG patients.
Hyperphagia
e introduction of high dose dexamethasone at the time of initiation of
radiotherapy is associated with an immediate increase in appetite. As a
consequence, children can show a dramatic weight gain within days and attempts
at controlling their appetite are often dicult because of the associated mood
swings and behavioural changes. e use of calorie-free drinks may help limit
the weight gain. When patients can be weaned o the steroids, the weight gain
is transient and most children go back to their baseline weight within weeks.
However, when steroids are continued, hyperphagia may lead to massive weight
gain that will limit even further the mobility of an already neurologically
handicapped patient.
Mood disturbance
e neuropsychiatric eects of steroids are probably the most common and
most stressful for parents and caregivers who often report that “their child is
not the same.” Steroids can cause anxiety, insomnia and irritability. Sometimes,
discrimination of these complications from manifestations of gliomas, cerebral
irradiation or changing intracranial pressure can be dicult in clinical practice,
and it is not uncommon that clinicians request a CT or MRI scan to rule out
complications such as intratumoral hemorrhage. However, steroids given at a
very high dose have without any doubt a signicant impact on behavior in some
children and negatively aect their quality of life. e management of these
neuropsychiatric side eects involves discontinuing or reducing the steroids as
much and as soon as possible. e use of neuroleptics can be considered on an
individual basis when discontinuation of steroids appears impossible.
Common Occasional Rare
Happens to 21-
100 children out
of every 100
Happens to 5-20
children out of
every 100
Happens to less
than 5 children out
of every 100
Early: within
days
• Hyperphagia
• Insomnia
• Gastritis
Prompt: within
2 to 4 weeks
• Immunosup-
pression
• Personality
changes
• Acne
• inning of
the skin
• Stretch
marks
• Weakness
• Infections
• Pancreatitis
• Increased
intraocular
pressure
• Hypertension
• Psychosis
• Vertigo
• Headache
• Spontaneous
fractures
• Growth sup-
pression
• Peptic ulcer
and gastro-
intestinal
bleeding
• aseptic ne-
crosis of the
femoral heads
Delayed: usu-
ally after several
months of treat-
ment
• Cataract(s)
Table 1: Side eects of Corticosteroids
Chapter 10: Use of Steroids in DIPG
144
145
Chapter 10: Use of Steroids in DIPG
Myopathy
Steroid myopathy is a complication that may signicantly impact the quality
of life of DIPG patients. e clinical manifestation of steroid myopathy is a
progressive weakness aecting mostly lower limbs that can have an impact on
walking abilities. Some children may require a wheelchair as a result of steroid
myopathy. Other consequences include the diculty to go up and down stairs
and an inability to run. Back pain is not exceptional and seems to be the result
of both myopathy and osteoporosis. Steroid myopathy usually improves when
the drug is discontinued or if the dose can be reduced. However, recovery can
take several months after steroid discontinuation. Not all patients will develop
steroid myopathy and the exact mechanism of this complication is unknown.
Some patients will present with severe symptoms of myopathy after 2 or 3 weeks
of steroid use, whereas other patients treated for months may have minimal or
no symptoms. It seems that regular exercise or physiotherapy programs may
help reduce the severity of myopathy.
Gastrointestinal bleeding and ulcers
Patients treated with corticosteroids are also usually treated with medications that
reduce the risk of gastric ulcer and hemorrhage. Although no signicant association
between steroid usage and gastrointestinal bleeding or ulcers has been identied in
children with brain tumors receiving steroids, it is prudent to use H2 antagonists
(like Pepcid) in DIPG patients treated with corticosteroids for a prolonged duration,
in particular when patients are treated with unusually high doses of corticosteroids.
However, in most patients treated with 1 or 2 mg. per day of dexamethasone, the
systematic use of H2 antagonists appears unfounded and twice-daily corticosteroid
dosing during meals reduces the risk of stomach irritation and spares the risks of side
eects and the expense of H2 antagonists.
Eects of corticosteroids on the immune system
Dexamethasone and other corticosteroids can cause immunosuppression by
inhibiting immune and inammatory responses and reducing the pool of
lymphocytes. e use of glucocorticoids will therefore increase the risk of
opportunistic infections. Pneumocystis carinii (PCP) is a fungal infection
responsible for life-threatening lung infections in immunocompromised
patients. ere is increasing evidence that patients with brain tumors receiving
high doses of steroids have an increased risk of PCP and in several DIPG studies,
cases of PCP have been reported as a result of the exclusive use of steroids
(without any concomitant chemotherapy). It is therefore recommended to
consider PCP prophylaxis when steroids are used for prolonged periods of time,
in particular when patients cannot be weaned o steroids.
In the context of progressive disease, swallowing disturbances can cause
signicant oro-pharyngeal stasis of secretions. e immunosuppressive eect
of the steroids will increase the risk of oral thrush (fungal infection) that can
be extremely painful and dicult to treat.
Alternatives to Corticosteroids
e large number of complications associated with prolonged or repeated use of
corticosteroids has led to the search for alternative therapies for the management
of peritumoral edema in brain tumors, and in particular in DIPG.
Xerecept® is a synthetic analog of the naturally occurring human peptide
corticotropin-releasing factor (CRF). Several animal studies have indicated the
ability of CRF to reduce the brain edema caused by brain tumors. Corticotropin-
releasing factor appears to reduce peritumoral edema by a direct eect on
blood vessels, independent of the release of adrenal steroids. Clinical trials have
shown promising activity against peritumoral edema in adult brain tumors. A
randomised trial has shown that Xerecept® benets patients with symptoms of
peritumoral edema associated with primary or metastatic cerebral tumors by
allowing them to reduce/stop their dexamethasone treatment, thereby reducing
the incidence of the steroid-related adverse eects of myopathy, cushingoid
symptoms, and skin disorders. A clinical trial is ongoing in children with
recurrent brain tumors (http://clinicaltrials.gov/ct2/show/NCT01369121).
Preliminary results suggest improvement in emotional, physical and fatigue
scores, and the possibility to reduce or even discontinue steroids.
Cyclooxygenase-2 inhibitors (like Celebrex) have been utilized by several
physicians for the management of progressive DIPG, either alone or in
combination with steroids. ere was indeed some suggestion that they might
be eective in treating cerebral edema. However, the cardiac complications of
this class of drugs have signicantly reduced the use of these agents in children.
Since VEGF plays an important role in the pathogenesis of peritumoral
edema, the use of inhibitors of VEGF, such as VEGF antibodies (for example
bevacizumab/tradename Avastin®) appears to be a logical option in the search for
alternatives to corticosteroids. In the context of DIPG a small study described
the ecacy of bevacizumab in children with DIPG with suspected radiation
necrosis. Four symptomatic children received bevacizumab for a period of 3
weeks to 3 months following completion of radiation at a dose of 10 mg/kg
every other week for a total of 3 to 6 infusions. Treatment was well tolerated
Chapter 10: Use of Steroids in DIPG
146
147
Chapter 10: Use of Steroids in DIPG
without evidence of side eects. ree of the 4 children were able to discontinue
steroids and had signicant clinical improvement in neurologic symptoms.
Further studies are planned to better delineate the role of this agent in the
management of children with DIPG.
Conclusion
Steroids and in particular dexamethasone have a major role in the management
of DIPG patients. However, due to the lack of prospective studies on the use of
steroids in this condition, our knowledge on the optimal dosing and schedule
of administration including weaning remains limited and most physicians
rely on their own experience when prescribing steroids in this context. ere
is currently signicant diversity in clinical practice and it is hoped that future
studies will provide more insight into the optimal use of steroids in DIPG, as
well as potential steroid alternatives.
Parent Perspectives
Initially you see the steroids as your saving grace. You hear the side effects
of extreme hunger and weight gain as possibilities but your pre DIPG
self knows that you can control your child’s portions and just encourage
healthier choices. There is no reason for significant weight gain for your
child. Sleeplessness won’t be a problem because your child sleeps like a rock.
Incontinence isn’t even mentioned in those early days. Muscle deterioration?
No problem, your child is active and strong. Anger management has never
been an issue at your house, tantrums are not acceptable.
After only a few short weeks you begin to find yourself thinking that since
your child is being put through all of this (until you get your miracle) he
or she deserves to eat mashed potatoes at all three meals and snack times
if he or she wants to. I mean carrots don’t really fill you up so what made
you think they would fill your child up? Slowly but surely over the course
of the next six weeks you find yourself looking at a completely changed
human being. The little person you took into the clinic on day one is not
the same person you are taking in some six weeks later.
It was difficult to see the effects of the Decadron on Andrew's six-year-old
body. His face was very full, and he did not look like himself. He saw his
reflection in the mirror a few weeks after diagnosis and commented, "My
cheeks are more puffy than they were." That was an understatement! The
weight gain was a direct result of his steroid-induced appetite and the
drastic decrease in his mobility. He liked to eat things that were good for
him, but he would sometimes cry because he was so hungry.
My daughter Hope was one of the children who (with the exception of 48
hours) was never off of the steroids. She gained over sixty pounds in just
six months and lost the use of her right arm. She was able to walk assisted
short distances until about five weeks before she died but other than that
was confined to a wheel chair. Her stretch marks were worse than any
Chapter 10: Use of Steroids in DIPG
148
149
Chapter 10: Use of Steroids in DIPG
pregnant woman I’ve ever met and eventually those marks became open
wounds in some areas. It was her great diligence and desire to persevere
that allowed her to wean off of steroids even if it was only for two days.
Hope was a hero.
In some respects, I felt that I lost Mara very soon after diagnosis. Her
personality was so markedly different that I felt the child I knew had already
left my home due to the steroids. The steroids make these kids turn into
something that they are not. That is our psychological horror as parents.
Oh, life with a child on steroids! Food becomes so important. Caleb is so
hungry all of the time. He told me yesterday that even though his tummy is
full, he still feels like he needs to eat. He cannot stop thinking about food.
Last night, he would not go to bed until he had two bananas on the bedside
table in case he woke in the night and needed a snack. My husband assured
him that we would be there and would get him something to eat if he needed
it. That wouldn't do. We had to have those bananas by the bed, ready at hand.
He awoke at about 6:00 a.m. and ate a banana. I helped him with that and
then fell back asleep. Every time I would drift off, Caleb would talk about
what he was planning to eat today. “When I get up, I am going to eat a
bowl of cereal and some scrambled eggs. Then I am going to go sit on the
back porch and eat a Pop Tart. Okay, Momma?” “Okay, Caleb,” I replied.
“Please just let Daddy and me sleep a little bit more and then we'll get
you some breakfast.”
Ten minutes passed, when Caleb said, “I want to be sure you pack my
lunch today when we go to the clinic. Okay, Momma? I want you to pack
spaghetti and meatballs, lasagna and a Caesar salad. Will you pack that
for me?” My reassuring reply was that we didn’t have to leave for the clinic
until noon, so he would be able to eat all of that before it was time to leave.
“Then can you be sure you pack a snack?” Content for five minutes that a
snack would be packed Caleb asked “Can we go to Krispy Kreme today?
We can get a hot and fresh for free and then get a dozen to bring home.”
…It's really quite comical from the outside, but I know he is just so tired
of it already.
From the day of diagnosis until the day he passed away, Bryce took steroids
to reduce the fluid surrounding the tumor. When we tried to take him off
of steroids after radiation, the headaches and nausea increased, so he
was always on a mild dose. We could tell the days when his cancer was on
his mind. He would become irritable and he would say, “This sucks, or I
hate this. Why does this have to happen to me? I don’t deserve this.” Our
response would always be a confirmation of his feelings. And it became
really difficult at times, because we knew that he was dealing with all of
these emotions, and so were we, but that we still had to parent.
Liam began Decadron immediately upon admission to the PICU however
we did not begin to see the side effects other than an increase in his
appetite until we returned home from the hospital. That’s when our love/
hate relationship began. Unfortunately Liam experienced many of the
difficult side effects from steroids. His appetite was massive. Ask anyone
whose child has had to take any significant amount of steroids and they
will tell you that eventually their child can turn any conversation, TV show,
game, whatever, into a conversation about food. Liam was no sooner half
way through breakfast when he was asking what was planned for lunch.
Fortunately for us and Liam during this time he craved only healthy things.
Fruit, vegetables (You’ve never seen a kid devour broccoli like our boy!),
seafood. He gained quite a bit of weight but thankfully not too, too much.
We were worried for a bit towards the end of radiation that his big cheeks
would not fir into his radiation mask and he would require a new one.
Thankfully though, that never happened. Although one would think having a
giant appetite would be the least of concerns when your child has cancer, it
is extremely difficult to see your child’s complete obsession with it. His love
of all things food though was not what was most difficult for Liam during
that time. Steroids brought huge mood changes, auditory hallucinations
that whispered horrible things to him. His anger was heartbreaking for us
to see, but also for Liam to feel. It sometimes made him lash out in ways he
never would have done before. He would cry after some of these moments
feeling awful that he may have hurt someone’s feelings, particularly mine.
Later when he would feel angry he would sit in his room and later when he
would call out to me, I knew he had settled down and was ready for a big
hug. Those were very difficult moments. Eventually Liam came to understand
Chapter 10: Use of Steroids in DIPG
150
151
Chapter 10: Use of Steroids in DIPG
that his medication would cause these moments and we all learned ways to
help him feel less out of control. As his symptoms improved, he was weaned
further and further down and eventually was able to completely come off
the steroids all together.
When Liam’s tumor began to progress, he was put back on steroids. That
was a difficult day. Liam however reassured me that it would all be ok. He
didn’t think he would have any of the troubles he had on the medication
previously and he was right! This time however he gained a huge amount
of weight. He became far less mobile in these months but the appetite
continued. We did our best to give him healthy foods to help combat the
lack of physical activity. Still he held a tremendous amount of weight for a
little guy who started out very small. His disposition however, was always
cheerful. Always. He did not have any of the problems with mood swings
and anger that he had previously. What a blessing!
Steroids do just what they are supposed to do in most cases. They help
alleviate swelling and really combat some of the scary symptoms that
children experience but they also come with some side effects that were
difficult to see. We tried our very best to help Liam manage those symptoms
as best we could. We talked a great deal about all of his medication and
their effects and we were careful to respect Liam’s feelings about how the
drugs were making him feel. After all it was his body, not ours. There were
several times when we asked to make adjustments whether it was changing
a dosage, changing a pill size, asking for a medication to be flavored or
crushed—whatever it took. There are many, many options available and
sometimes it’s just a matter of asking the question of your child’s provider.
If your child has difficulty swallowing pills, is that medication available
in a suspension? Always ask the question. Your child being diagnosed with
cancer presents parents with the steepest learning curve they will ever
encounter. Never, ever be afraid to ask questions!
The neuro-oncologist discussed some of their primary concerns going
forward. He indicated that they were concerned about the level of
hydrocephalus (an accumulation of cerebrospinal fluid on her brain) and
the impact that may have on her functioning. He also recommended that
Stella be taken off the steroids. While the steroids control the effect of
the hydrocephalus, they also caused her to have a significantly increased
appetite and to be agitated and angry. These side effects of the steroids
are problematic since one of the most common symptoms of the tumor is
difficulty swallowing. There is a concern that should that occur while she
is on the steroids, the anger that may result from the hunger and inability
to eat may cause choking.
When Caleb was first diagnosed with DIPG he was started on steroids. At
that time he took them for about three weeks during the first half of radiation.
The dose was fairly small and it helped with his speech and his balance.
When he first started them his initial side effect was extreme hunger and
after about a week we began noticing the mood swings. I remember the first
time I really saw what the steroids could do to his personality. We had taken
him bowling. He was trying so hard and wanted to do well but it just wasn't
going his way. He became very agitated and I can remember thinking that
this was not my child. He was 8 years old at the time and although he had
always had a competitive streak, he was very even tempered. Here he was
having a mini tantrum after each throw of the ball. I was horrified thinking
I would never see my sweet natured boy again. Of course as the steroids
were weaned he became more and more like himself. I was so relieved and
truly thought I never wanted to put him on that wretched drug again—he
no longer looked the same and at times he certainly didn't act the same.
Once he was off the steroids he stayed off them for two months. We had
learned of tumor progression the month before and had opted for a second
round of radiation. He was nearly finished the second round when one
morning his whole left side became paralyzed and he could barely speak.
We took him to the hospital and they started him on a fairly high dose of
dexamethasone again. I remember thinking it would just be temporary
and I wouldn't agree to long term use. We did a slow wean but as we got
closer to being off he would begin with symptoms again. We were being
told the symptoms were likely due to swelling from the radiation and as the
swelling came down so would the dexamethasone, so we would increase
again while we waited for the swelling to go back down. This went on for
months... wean the “dex,” increase the “dex,” wean the “dex,” increase
the “dex,” and each time we needed to increase the dose, he would need
a bigger increase to get him back to baseline.
At this point, it was thought the symptoms were likely caused by radiation
necrosis. Caleb was so hungry and became agitated and food obsessed, he
gained over 36 lbs. in 3 months and the weight gain continued. I remember
Chapter 10: Use of Steroids in DIPG
152
him trying to control it. His moods were crazy at times, thankfully these
bouts of insanity were usually short lived but his mood swings would
upset him. He would feel so badly for acting irrational and no amount of
reassurance would make him feel better about it. He knew right from wrong
and sometimes the steroid would make him behave wrong. It was our double
edged sword. On one hand it helped the tumor symptoms but at what cost
to his quality of life? I feared if we lowered the steroids completely we
would lose him faster, I wasn't ready, I knew I never would be.
So, we started Avastin, which was thought to possibly reduce the
effects of radiation necrosis, in hopes that we would be able to stop the
dexamethasone. I am not sure if it helped. I think it might have, as we were
able to lower the dose but at this point in time no amount of the steroid
would help him to walk again, He was in his wheelchair full time, had no
use of his left side, could no longer swallow liquids or solids that hadn't
been pureed, and his speech was greatly affected. On top of all that his
skin had been stretched beyond its capabilities and he had open wounds
on his body. Thankfully most of them caused no pain but he had two, one
under his arm and the other between his bum cheeks that caused him a lot
of pain. We were able to get topical morphine for the wounds which helped
a lot. At this point we knew the steroid was causing more harm than good
so we did a fairly rapid wean.
Caleb took his last dose of steroid on a Tuesday and joined the angels the
following Saturday. Through it all, Caleb had tons of beautiful moments
of pure joy, days when he was all smiles and would enjoy the company of
those around him. He planned things and wanted to do them and it was
days like this which made the steroid decision so hard for us. I know the
steroids caused him a lot of anguish especially in the end but I also know
it gave us a lot of beautiful, happy, loving days.

153
Chapter 11: Caring for Your Child at Home
Chapter 11
Caring for Your Child at
Home
Deborah Lafond, DNP, PNP-BC, CPON, CHPPN
If your child has been diagnosed with diuse intrinsic pontine glioma (DIPG),
your family will inevitably face some challenges while caring for him or her at
home. For example, your child may have trouble taking or tolerating medicines
or have problems with swallowing/eating, walking, sleeping, and other aspects
of daily living. Many parents feel anxious about taking their child home after
treatment. Figuring out what your child needs and how to care for your child at
home can be very stressful.
You may have spoken with other parents of children with DIPGs and may have
heard about some challenging situations. Although it is dicult, try not to
compare your experience with any other child and family. Each child and family
is special and each situation is unique.
To prepare you for caring for your child at home, this chapter will describe
common problems you might encounter and oer advice about how you and
your child can best manage these issues.
Your Child’s Health Care Team is Available to Help You
Remember that your child’s health care team is there to help you and your child cope
with the challenges of DIPG. Your family may not encounter any of the challenges
listed in this chapter, or you may experience only some of them. While the health
care team tries to anticipate what you and your child might need, not all situations
are easy to predict. Don’t hesitate to contact your child's health care team whenever
you have questions or concerns.
Know whom to call
You have likely met many members of
your child's health care team since the
Dr. Lafond is a Nurse
Practitioner in Neuro-
oncology and Palliative Care
at Children’s National Medical
Center, Washington, DC.
Chapter 11: Caring for Your Child at Home
154
155
Chapter 11: Caring for Your Child at Home
time your child was diagnosed with a DIPG. Each person has a special role
in caring for your child. Make sure you have the name, phone number, and
email contact information for the person on your child's health care team
that you should contact for routine questions and concerns. Also, be sure
you have the contact information for those you should contact during an
emergency, after hours, and on weekends. e American Childhood Cancer
Organization provides a free journal entitled Along the Way to assist with this
type of record keeping. is journal is available without charge by request
to all parents of children with cancer. is journal includes designated pages
to include contact information for your child’s healthcare team. Keep the
contact information:
• Next to every phone in your house.
• In your cell phone. (If you own an iPhone, there is a helpful “app” called
iCANcer, available through iTunes, where you can store all contact
information, as well as the diagnosis, treatment, and medical information
of your child.)
• In your wallet or purse.
• Near your phone at work (if you have a private oce or desk space).
• At your child's school—with your child's teacher and the school nurse
and at your other children’s school(s), if applicable.
• With people who care for your child, either at your home or theirs, while
you are gone (such as grandparents, daycare, or babysitters).
Your child's social worker
Get to know your child's social worker well. e social worker is there to help you
and your child navigate the many feelings and emotions you might experience as
you go through the DIPG journey. If you have not been assigned a social worker,
call your child's nurse and ask to have a social worker assigned to your family.
Medical problems are not the only issue you, your child, and the rest of your
family will face. Having a cancer diagnosis is a scary and emotional experience.
ere will be times when you are sad, times when you are angry, times when you
are frustrated, and times when you feel joyful. Your social worker can help you
and your family as you deal with these dicult emotions.
Your child’s nurse or nurse practitioner
In general, social workers cannot answer medical questions about your child's
diagnosis and treatment. us, your child’s nurse is another important contact
on your child's medical team. Your child will likely have a primary nurse (RN)
or nurse practitioner (APN) who will take the lead in answering all your medical
questions. e role of APNs is similar to the role of doctors; they can serve as a
patient’s primary health care provider and prescribe medications, whereas RNs
do not. e nurse/nurse practitioner is usually the rst person you call with
routine questions and concerns. If that person does not know the answer, she
or he can readily nd your child's doctor. is does NOT mean your child's
doctor is not routinely available to you. You may talk with your child’s doctor
at most any time, but your child's nurse/nurse practitioner is a great resource
for practical information about the day-to-day challenges you may face. Do not
hesitate to call the nurse/nurse practitioner if you have questions or concerns.
e nurse/nurse practitioner usually serves as a central contact for parents and
helps get them to the right person when questions or concerns arise.
Your child’s case manager
Another key person to get to know is the case manager assigned to your child
from your health insurance company. If a case manager is not assigned to you,
call your insurance company and ask to be assigned one. Tell the insurance
company that your child has “a brain tumor with complex health care needs;”
this is common terminology you can use to help your insurance company
understand why you are asking for a case manager. If your insurance company
is not responsive, be persistent.
You will need the following information when you talk with your insurance
company. is is information that you can get from your child's nurse/nurse
practitioner.
• e diagnosis: diuse intrinsic pontine glioma (ICD-9 code is 191.7);
• Date of diagnosis;
• e name of all hospitals where your child receives treatment;
• e type of treatments your child is receiving, (e.g., chemotherapy,
radiation therapy, physical therapy, occupational therapy);
• The contact information for your child's doctor and nurse/nurse
practitioner.
Be willing to give your insurance company permission to contact your child's
doctor and/or nurse/nurse practitioner for more information about your child
and the care your child will need. e insurance company may have other
questions regarding your child's diagnosis. Do not hesitate to ask the insurance
Chapter 11: Caring for Your Child at Home
156
157
Chapter 11: Caring for Your Child at Home
representative to contact your child's doctor or nurse/nurse practitioner to
get answers to these questions, especially if you do not know the answers or
are unsure of the details.
Having a case manager at your insurance company usually allows you to have
a central contact person who can help you obtain referrals and approvals for
your child’s care. By having one person (your case manager) to call for help
with insurance-related issues, you can often avoid the long process of trying
to nd the right department for needed help. Your case manager may also be
able to arrange for someone to help care for your child at home.
Home care nurses
Sometimes parents can arrange nursing care in the home for a short while.
is is not possible in every situation, but it may be possible for short periods
of time. Talk with your child's case manager, social worker, or nurse/nurse
practitioner to nd out whether or not you are able to have home care nurses
come in for short periods to help you, especially when your child rst goes
home or if your child's symptoms worse
n and he/she requires more care at home.
General Physical Care
Every child is unique in the symptoms and care needs they will have at home.
Your child may have none of the needs mentioned here, some of them, or all
of them at any given time during the DIPG journey. At times, it may feel as
if you have to be your child's at-home nurse. It is important to know what to
expect and how to provide care for your child so he/she can stay at home and
out of the hospital as often as possible.
is caretaking can be overwhelming at times, but the goal is for your child
to live as normal a life as possible, doing the things that he/she enjoys, such
as playing with friends, going to school, and being with family. Knowing how
to care for your child may help prevent some of the most common problems
or help resolve them sooner. is section covers such issues as prevention,
medications, breathing problems, nutrition/feeding issues, constipation, skin
breakdown, nausea/vomiting, pain, mobility issues, sleeping, caring for central
lines, and some common symptoms children with DIPG experience.
Prevention
Discuss with your health care team any potential problems your child might
have. Ask about what you may need at home to care for your child. Be sure
that you understand what complications the health care team thinks your child
may experience, what medications they are sending home, and the possible
side eects of these medications. Also, ask your health care team what signs of
medical problems to look for if your child's condition changes.
Infection
Infection can be a serious risk for your child, especially if he/she is receiving
steroids, such as dexamethasone or certain chemotherapy agents. Being
on certain other medications can also make your child more susceptible to
infections. Having an infection can cause serious complications for your child,
so ask your doctor or nurse/nurse practitioner if your child is at increased risk
of developing infections. Infections can be bacterial, viral, or fungal.
You don’t need to keep your child away from other people unless someone is ill.
You can let your child go to school or play with other children. But if another
child is obviously ill, it is best not to be around that child until he/she is well.
If other family members are ill, it is often dicult to keep your child separated
from them, and it is not recommended that you keep your child in another
room or house. Just try to be as careful as possible about close contact.
e most important way to prevent infection is to have good hand washing
practices. You may use regular soap, wash for at least 30 seconds and have
several bottles of alcohol-based hand sanitizers available in your home and
small ones to keep in your purse, car, and your child's backpack or diaper bag.
Teach your child to wash his/her hands well (including under the ngernails)
after using the bathroom, before and after eating, after playing with pets, after
touching other people, or anytime his/her hands are dirty. Also ask other people
including other children who are in contact with your child to wash their hands
well before they interact with him/her.
Medications
Ask the nurse how to give your child his/her medications and at what times
each one should be given. e nurse can make a schedule of all medications
with a place for you to record the day and time you give the medications; this
type of form is helpful when your child takes several medications. Having a
schedule and a record of when you gave each medication will help you remember
when to give them to your child. A sample medication form is included in
Appendix A. An online medication program is available at: http://www.
medactionplan.com/medactionplan/mymedschedule.asp.
Ask your doctor or nurse practitioner the following questions about each
medication your child has been prescribed.
Chapter 11: Caring for Your Child at Home
158
159
Chapter 11: Caring for Your Child at Home
• Why has this medication been prescribed (why it is needed)?
• When should I give this medication to my child (what schedule)?
• How should I give this medication to my child (with or without food)?
• If pills are prescribed, can they be split or crushed for easier swallowing?
Do not crush pills or dissolve pills or capsules in liquid without talking to
your pharmacist or nurse/nurse practitioner rst. Some medications cannot
be safely crushed or dissolved.
• What are the side eects of this medication and what can I do to manage
or prevent them?
• Are there any other medications, including over-the-counter medications,
that I should avoid giving my child?
• Are there any foods that I should avoid giving my child that might interact
with the medications?
• What should I do if I forget to give a dose of a medication or if my child
vomits up the dose/pill?
If your child has any diculty taking the medications or experiences side
eects, let your doctor or nurse practitioner know as soon as possible so they
can advise you about what to do. Not all medicines have unpleasant side eects
or interactions with other medicines. Hopefully, your child will have no side
eects with the medications he/she is taking, but it is best to be prepared for
what to expect, possible side eects, and how to deal with any that may occur.
Additional tips for medications:
• Keep all medications in a safe place, out of reach of children.
• Record the time you give each medication on the daily schedule and keep
track of any side eects your child experiences. Keep this list up to date
and bring it with you to every doctor’s appointment or emergency room
visit so everyone knows exactly what medications your child is taking and
the last time he had each one. is information can be documented in the
iCANcer app if you own an iPhone, iPod Touch or iPad.
• Keep medications in a cool, dry, place, especially if the weather is hot
and humid. Do not refrigerate medications that are not supposed to be
refrigerated, but do not leave medicines in direct sunlight or in a hot car.
• If your child has trouble swallowing pills or liquid medications, ask your
nurse for help in nding the best way to give them. For example, some
medications can be mixed with special avors at the pharmacy to make them
taste better or can be crushed and put into pudding, ice cream, chocolate
syrup, or applesauce.
• For older children and teenagers, consider getting a pillbox to organize
medications to be given at certain times of the day. Most drug stores and
large grocery store chains sell pillboxes in one day/time or multiple day/
time versions.
• Measure all liquid medications with a syringe, if possible, rather than a
teaspoon. Regular kitchen spoons can be dierent sizes and you want to
make sure you give the right amount of medicine each time.
• Make sure you understand each medication and why it is being prescribed.
In addition to your doctor or nurse/nurse practitioner, your pharmacist
can provide you with information about each medication. If you use the
Internet to nd information, please double check that information with
your pharmacist, doctor, or nurse/nurse practitioner to ensure you get
accurate information.
• Do not use medications that have not been specically prescribed for your
child. And do not share your child’s medicines with anyone else, even if
that person takes the same prescription.
• Do not give your child any herbal or vitamin medications without checking
with your doctor or nurse/nurse practitioner. Some common herbs and
vitamins can interfere with chemotherapy and radiation treatments, as
well as possibly interact with other medications your child is taking. More
information about complementary and alternative medicines (CAM) is
discussed in a section later in this chapter.
Pain
Pain can occur in children of any age and at any time. ere are dierent types of
pain depending upon the cause, the location, your child's special characteristics,
the treatments your child is receiving, and your child's past experiences with
pain. Not every child with DIPG will experience pain. Some children feel no
pain at all. If your child does experience pain, the goal is to relieve that pain
as quickly as possible, with the least amount of side eects. is section will
describe some ways you can help your child manage pain.
It is important to be able to recognize when your child is in pain. Your child
will experience the normal aches and pains of childhood, but sometimes it can

Chapter 11: Caring for Your Child at Home
160
161
Chapter 11: Caring for Your Child at Home
be dicult to gure out whether a complaint of pain is a normal childhood
experience, such as bumps and bruises from play, or something more serious.
Depending on the age of your child, it can be dicult to gure out whether
your child is even experiencing pain. For example, infants normally cry, so it
can be hard to determine if a cry is from pain or something else. Older children
and adolescents may not admit to feeling pain because they may associate
pain with having to go to the hospital or taking more medications, which
may make them sleepy or nauseated. If you think your child might be in pain,
please talk with your doctor or nurse/nurse practitioner about ways to manage
your child's pain. Remember that while pain is physical, it can also have an
emotional component. Each child reacts dierently to pain, so it is important
to understand how your child expresses pain. Indications that your child may
be in pain include:
• A high-pitched cry or change in an infant's normal cry;
• Changes in facial expressions;
• Rubbing particular areas on the body that may indicate pain;
• Irritability or restlessness;
• Being inconsolable;
• Being less active or mobile than usual and playing less;
• Loss of appetite or changes in eating patterns;
• Changes in sleep patterns.
If your child can talk, ask him/her if he/she has pain and ask him/her to show
you where the pain is, what it feels like (e.g., sharp, stabbing, shooting, burning,
cramping), what he/she thinks is causing it, and what he/she thinks will help
it feel better. You may also choose to show your child pictures of faces that
represent no pain to severe pain and ask him/her to point to the picture that
best represents how he/she is feeling [Fig. 1]. e answers to these questions
can help you decide how to help your child.
Figure 1: Faces Pain Scale, used with permission by the International Association for the
Study of Pain (IASP)
Ways to treat pain without using medications include:
• Reassure your child that pain does not always mean the DIPG is getting
worse; explain that all children experience pains during childhood.
• Create a calm and nurturing environment by turning down any bright
lights, minimizing noise, and creating a comfortable room temperature.
Experiment to nd out what works the best to calm your child.
• Use distraction (e.g., singing, reading books, blowing bubbles, telling stories,
watching a favorite movie DVD, etc.), relaxation techniques, visual imagery,
or play to get your child's mind o his/her pain. Ask your child life specialist,
social worker, or psychologist for other strategies to help your child.
• Apply a heat pad or ice pack, whichever your child prefers, to areas of pain.
• Run a warm bath or shower to help your child relax.
• Use massage or gentle touch, but only if your child wants to be touched,
because sometimes touch is not comforting. Massaging your child’s hands,
feet, and shoulders can help your child relax and give you a way to connect
with your child. Many hospitals oer massage classes to teach you how to
perform massage techniques eectively.
• Play music—soothing music is best but use music that your child enjoys;
or try a sound machine with sounds such as running water or ocean waves.
• Have your child's favorite blankets or stued animals or other favorite toys
readily available.
• Hold your child to cuddle or rock him/her, or lie in bed next to him/her
(if he/she wants you to, as older children may like to be left alone to sleep).
• Let your child cuddle up with the family pet.
Ways to manage pain with medications include:
• In general, start with milder medicines such as acetaminophen or ibuprofen.
However, these medicines cannot be given with certain chemotherapy
medications or when blood counts are low, so always check with your child’s
doctor or nurse practitioner before giving your child any over-the-counter
pain medications.
• With prescription pain medications, read the directions carefully. ese
medications are prescribed based on your child's age and weight. Do not
give more than the recommended dose unless you are told to do so by your
Chapter 11: Caring for Your Child at Home
162
163
Chapter 11: Caring for Your Child at Home
child's doctor or nurse practitioner.
• If your child has more moderate to severe pain, your doctor or nurse
practitioner may prescribe stronger medicines, such as opiates (morphine,
codeine, methadone, etc.). Many types of opioids can be safely used for
children. Your doctor or nurse practitioner will explain the directions for
giving opioid medications and the expected side eects.
• In general, it is best to give pain medications throughout the day on a regular
schedule, or when your child’s doctor or nurse practitioner tells you to give
them. Giving medicines on a regular schedule avoids the peaks and valleys
that occur when they are only given when your child experiences pain. Just
be sure not to give your child more medication than recommended by the
doctor or nurse practitioner, or more than is recommended on the bottle
for over-the-counter medications.
• If your child is experiencing increasing pain, even though he/she is on
strong pain medication, other medicines can be added to help the pain
medicines work better.
• Every child is unique, so the plan for managing your child's pain should
be the one that works best for him/her.
Common concerns about pain medicines include:
• Addiction and tolerance: Children who are in pain may need strong
medications to help relieve it. If your child needs pain medications for a
long period of time, he/she may need increasing doses of pain medicines
to help relieve the pain. is need for higher dose is known as tolerance,
meaning your child needs a higher dose to get the same eect. Tolerance is
NOT the same thing as addiction. Addiction is a physical and psychological
craving for medication, usually to achieve a "high." Children in pain do
not become addicted to pain medications if they are used appropriately and
only as your doctor or nurse practitioner tells you to give them.
• Medicine is too strong: Parents may worry that if a strong pain medication,
such as morphine, is prescribed it means their child is getting worse.
However, morphine is a very good pain medicine and works well for
children of all ages, from infants to adolescents. Morphine has been used
for many years and doctors and nurse practitioners know a lot about
this medication. ere are other pain medications that can be used, but
morphine is frequently used because it relieves pain so well. If the doctor
or nurse practitioner prescribes morphine, or a similar opioid medicine, it
does not necessarily mean your child's condition is getting worse. Don't
be afraid to ask your doctor or nurse practitioner why a particular pain
medication is being prescribed.
• Multiple pain medications: Sometimes medicines may not work too well
by themselves, but in combination with other medicines work very well.
You may even be able to use smaller doses of each medication when they
are combined. Sometimes pain has several dierent causes, so one pain
medication does not treat all the causes. Talk with your doctor or nurse
practitioner about why your child is taking each medication and ask them
if you can eliminate any of the medicines.
• Side eects: If your child is taking opioid medications, be sure to start
him on a stool softener to prevent constipation. Opioid medicines slow
down the intestines, which can cause constipation. Itching, nausea, and/or
increased sleepiness may also be caused by certain medicines. Be sure to tell
your doctor or nurse practitioner if your child has any of these symptoms
so you can get advice about how to handle the side eects. Generally these
types of symptoms happen in the rst few days that your child is taking
opioid medicines. Talk to your doctor about any other side eects that you
think your child might be experiencing from other pain medications as well.
Nutrition and Feeding Issues
Good nutrition is important for your child’s growth and general health. If your
child is having issues with feeding/eating, it is important to nd out why. For
example, if your child is nauseated from chemotherapy, medicines can be given
to help calm his/her stomach. If your child is too tired to eat, nding ways to
deal with fatigue might help him/her have times when he/she feels better and can
eat. Having a meal schedule might also help your child eat regularly, as multiple
doctor appointments, other family obligations, and the chaos of daily life can be
overwhelming and cause a child to lose his/her appetite or forget to eat.
If family members and friends have asked how they can help, consider letting
them make meals for your family so you can concentrate on caring for your child.
Your child’s illness may change over time, and there may be times when he/she
wants to eat more or eat less. ings that will aect nutrition and the ability to
eat for a child with DIPG include the following:
• Weight gain: Your child may gain weight rapidly when he/she is on steroids.
You may notice that he/she has a ravenous appetite often way out of
proportion to what he/she normally eats. It can be very dicult to control your
Chapter 11: Caring for Your Child at Home
164
165
Chapter 11: Caring for Your Child at Home
child’s appetite, but some tips include limiting high-calorie foods and salty
foods. is can be dicult, because your child may want to eat only certain
foods. Let him/her eat his favorite foods but in moderation. For example,
you may try saying something like, “You can have the chocolate ice cream one
time today. If you eat it now, then you cannot have any more until tomorrow.
Are you sure you want to eat it now, or would you rather try _______ now and
save the chocolate ice cream until later today?” is approach does not always
work because children often live in the present moment and to them "later"
really does not exist; later means never! You can also try a sticker chart or
some reward system so your child has to eat certain “good/healthy” foods in
order to earn the privilege of eating the less-healthy foods. is can be a real
challenge, so don’t hesitate to talk with your child’s social worker, nurse, or
child life specialist about tips to help you manage your child’s food cravings.
Consulting with a nutritionist or dietitian may be helpful for learning ways
to decrease calories in your child’s diet.
• Weight loss: On the opposite side of the spectrum, your child may lose
weight. is might happen while your child is getting certain chemotherapy
medicines or radiation therapy, or when being weaned o steroid medicines.
As frustrating as it may be to have your child eating all the time, many parents
nd it as frustrating to see their child not eating and losing weight. ings that
can help counteract weight loss are using supplements, such as Pediasure©,
Boost©, or Carnation Instant Breakfast©, or other supplements that your
doctor or nurse practitioner recommends. Your health care team will tell you
which supplements they recommend and how much to give your child each
day. Consulting with a nutritionist or dietitian may be helpful for gaining
tips about increasing calories in your child’s diet. ere are also medicines
that can be used to stimulate your child’s appetite, but before using any you
should ask your health care team if they think an appetite stimulant would
be helpful for your child. Also remember that if your child is tired and less
active, his/her metabolism slows down and he/she does not need as many
calories. Your child may also have cravings for a certain food and then only
take one or two bites. is can be quite frustrating for parents. Try to let
your child eat and drink what he/she wants. Sitting down to a big plate of
food or large glass of a liquid beverage can be overwhelming for a child. Try
to oer small, frequent meals. You will be surprised by how the calories add
up by just taking a few bites many times a day!
• Diculty swallowing: Many children with DIPG have diculty swallowing.
is is most common with thin liquids as they move more quickly through
the mouth and throat which can result in aspiration, but can happen with
any food or liquid. If your child coughs or chokes when he/she eats, it may
be a sign that he/she is having trouble swallowing. If your child has diculty
swallowing solid foods, try cutting food up into very small bite-sized pieces,
serving soft foods, and encouraging him/her to chew food thoroughly and
not rush while eating. If your child has diculty swallowing thin liquids,
try using a syringe to get a stream of liquid down, using thickening agents
to bulk up thin liquids, or using thicker versions of similar liquids
(e.g., milk
shakes or liquid yogurts instead of milk, and applesauce instead of apple juice)
.
• Inability to eat by mouth: ere may come a time when your child cannot
take food or liquids by mouth, so this is a challenge to think about in advance.
Ask your child’s doctor or nurse practitioner about the dierent causes of the
inability to eat or drink. Knowing about possible causes will help you notice
signs and make decisions about how to help your child if that time comes.
If your child reaches this point, the doctor or nurse practitioner will conduct
a physical exam to nd out the cause of the problem. If it is a mechanical
problem, such as the vocal cords not closing properly or aspiration (when food
goes down to the lungs instead of the stomach), then maybe your child can
receive uids and nutrition through an alternate method, such as a nasogastric
tube (NG-tube) or gastrostomy tube (G-tube). If the DIPG is quite advanced
and your doctor or nurse practitioner thinks your child is nearing the end
of life, then putting your child through the discomfort of having a tube
placed to receive uids and nutrition may not be the decision you want to
make. If your child’s DIPG is advanced, he/she may not want to eat as his/
her metabolism and digestive system slow down. It is not uncommon for the
appetite to signicantly decrease as a child’s illness becomes more advanced.
More about these dicult decisions will be discussed later in this chapter.
• Articial uids and nutrition: is is the term the health care team uses
for giving uids and nutrition through a nasogastric tube (NG-tube) or
gastrostomy tube (G-tube). e term “articial” simply means that the
uids are given through a tube rather than orally. An NG-tube is a small
exible tube inserted through the nose, down the back of the throat, and
into the stomach. e tube can stay in from a few weeks to a few months,
and it is used as a temporary way of giving uids and nutrition. If your child
has problems with nausea or vomiting, he/she may vomit up the tube and
the tube may have to be replaced more often. A G-tube is a bit larger and is
placed directly into the stomach or intestines by an interventional radiologist,
gastroenterologist, or surgeon, usually while your child is under anesthesia or
sedation. A G-tube can stay in for about 6 months but can be easily changed
by a home care nurse or—in some cases—you may be taught how to change
Chapter 11: Caring for Your Child at Home
166
167
Chapter 11: Caring for Your Child at Home
the tube. Either an NG-tube or G-tube can be hooked up to a feeding bag to
give liquid nutrition through either several bolus (large) feedings at dierent
times during the day or by a small amount being dripped in with a feeding
pump over 10 to 12 hours at night. Dierent types of liquid feeding formulas
are used depending upon the nutritional needs of your child. Your child's
nurse or nutritionist will go over the best type of schedule for your child and
explain how to manage these feedings at home.
ese types of feedings are not without complications, so your doctor or nurse
practitioner should review the risks and benets with you so you can make
an informed decision. In general, if the benets are great and the risks are
small, a feeding tube might make a lot of sense. If your child is at great risk for
complications from a feeding tube (for example, if he/she is not able to process
and eliminate the feedings, causing swelling and uid accumulation in the lungs)
then a feeding tube may not be the best choice. Having open, honest discussions
with your health care team will help you to make these decisions.
Nausea and vomiting
Nausea (feeling sick to your stomach) and/or vomiting can occur for many
reasons. Your child may have initially had nausea and/or vomiting as one of the
rst symptoms of his/her DIPG. Usually this is due to increased intracranial
pressure caused by swelling and pressure on sensitive areas of the brain. To
combat sickness, medications such as steroids can help decrease swelling in the
brain. Other things that can cause nausea and/or vomiting include constipation,
uid and electrolyte imbalances, chemotherapy, and certain medications (such
as opioids). Tips for decreasing nausea/vomiting include:
• Giving anti-nausea medications, such as Ondansetron or others that your
doctor or nurse practitioner might prescribe for your child, about 30
minutes before chemotherapy and/or radiation treatments and as instructed
at other times of day, as needed.
• Giving your child's medications with food or after a meal, depending on
the medication.
• Feeding your child small, frequent meals.
• Giving your child smaller amounts of liquids with meals and avoiding
carbonated beverages.
• Avoiding greasy, fatty foods.
• Avoiding foods with strong odors.
• Putting a drop of peppermint oil or other strong-avored oil of your child's
preference (available at your local nutrition store and some pharmacies) on
your child's upper lip, just below the nose.
• Using aromatherapy (described in more detail in the section below about
complementary and alternative therapy).
• Taking your child outside to be in the fresh air or having your child sit
near a fan.
Constipation
Constipation, or diculty having a bowel movement, can cause discomfort
for your child and lead to pain, nausea, vomiting, decreased appetite, and
irritability. Constipation is generally dened as not having a bowel movement
in more than 3 days, pain or crying with passing stool, or the inability to pass
a stool after 10 minutes or more of trying. You may notice that your child's
bowel movements are hard or small pellets, which are also signs of constipation.
Constipation is a common problem in childhood and can be even more of a
problem for a child with DIPG when taking certain medicines or when the
tumor involves the spine. Track your child’s normal bowel movement routine.
For example, do they usually have a bowel movement every day, several times
per day, every other day, etc.? If your child does not have a bowel movement
per his/her usual routine or you notice that the stools are hard or dicult to
pass, contact your child’s doctor for advice.
Several things can help prevent constipation:
• Have your child drink plenty of uids, especially water.
• Increase ber in your child’s diet and encourage him/her to eat fresh fruits
and vegetables.
• Increase your child’s physical activity.
• Establish a bowel regimen (i.e. having your child sit on the toilet for 5–10
minutes after meals and at bedtime).
• Consider giving your child a stool softener or stimulant (laxative), but rst
talk with your child’s doctor or nurse practitioner for advice about which
type is best for your child.
• Avoid suppositories or enemas unless recommended by your child’s doctor
or nurse practitioner.
Chapter 11: Caring for Your Child at Home
168
169
Chapter 11: Caring for Your Child at Home
Skin Issues
Skin problems are common in children with DIPG and are usually related to taking
steroid medicines (e.g., dexamethasone). Common skin problems include striae
(stretch marks) and dry skin. In addition, some children with decreased activity and/
or decreased mobility may be prone to developing pressure sores. Common places
for pressure sores to develop are on the lower back, buttocks, hips, or heels. Striae
can develop anywhere on the skin surface.
Some tips to help manage skin problems in your child include:
• Changing your child’s position frequently to relieve pressure on any one area (if
your child’s mobility is limited).
• Bathing your child daily and looking carefully at the skin for changes.
• Using moisturizing lotions/creams. (Be careful about this if your child is receiving
radiation therapy. First talk with the radiation doctors.)
• Using sunscreen (SPF 30 or higher) when outside, even if your child has a dark
complexion.
• Having your child drink plenty of uids to stay well hydrated.
• Using extra padding on the bed or wheelchair.
Fatigue
Many children with DIPG feel very tired from time to time, which can be
distressing for the child and family. Children want to be able to play, go to
school, and do all the things they used to enjoy doing. Fatigue (feeling very tired)
can be caused by muscle weakness, low blood counts, nutritional deciencies,
stress, and certain medications or treatments, especially radiation therapy.
Many children experience a great deal of fatigue during and for a few weeks
after radiation therapy; this is called radiation somnolence syndrome. Ask
your doctor or radiation oncologist to explain this condition. It goes away by
itself and usually does not require any treatment.
ings you can do to help your child if they have chronic fatigue include:
• Planning important activities for the time of day when your child is usually
most awake.
• Giving your child pain medicines or using some of the non-medicine
methods of managing pain, as pain is often a cause of fatigue.
• Making sure your child gets adequate sleep/rest; consider putting a "DO
NOT DISTURB" sign on the door when your child is resting during the
day.
• Limiting visitors to allow for rest time.
• Letting your child go to school for just one class, or half a day if he/she can;
consider home schooling or a tutor (if your child is unable to go to school
for a full day). Also, your social worker can help arrange for a home tutor
from the school system.
• Talking with your doctor or nurse practitioner to nd out if using a
stimulant medication would be helpful for your child. ere may be
stimulant medications that can be used if the other methods do not help.
Some children become more fatigued as their disease becomes more
advanced, often sleeping many hours a day. is can be scary for parents
and other family members. Talk with your health care team about this.
Allow your child to rest, but reassure your child that you are never far away
and that you will be there when needed.
Breathing Problems
Some children with DIPG have diculty breathing. is issue can be due to
increased secretions (drooling) in the mouth and back of the throat, weight
gain associated with use of steroid medications, infection, coughing, wheezing,
low blood counts, or the tumor aecting the nerves that control breathing. It
can be scary for you and your child if he/she has trouble breathing. One of the
most important things you can do when your child has trouble breathing is
to remain as calm as possible and help your child remain calm, as anxiety may
increase breathing diculties.
Here are some tips to counteract common causes of breathing diculties for
children with DIPG:
• Reduce anxiety: Create a calm environment. Even though the moment
may be frightening, take some deep breaths to calm yourself down so you
can remain calm and in control. Ask your child’s social worker or child life
specialist to teach you techniques to help calm your child down. Sometimes
playing soft, quiet music, speaking in a slow calm voice, and doing some
deep breathing exercises can help your child calm down and relax so he/
she is not as short of breath.
• Combat shortness of breath: Teach your child to take slow, deep breaths
Chapter 11: Caring for Your Child at Home
170
171
Chapter 11: Caring for Your Child at Home
when he/she starts to feel short of breath. Other tips include using a small,
hand held fan to blow air gently in your child’s face; keeping the room well
ventilated or opening a window; changing your child’s position—such as
from lying to sitting, to lying on his/her side, to elevating the head of the
bed or using another pillow to raise up his/her head and shoulders.
• Avoid smoke: Do not smoke around your child and don’t let other people
smoke around your child.
• Reduce increased secretions: Your child may have diculty swallowing
his/her saliva, or may make noisy or gurgling sounds when he/she breathes.
is is usually due to saliva pooling in the back of the throat. You may also
notice that your child is drooling more from his/her mouth. ere are some
medicines that can be used to help dry up the saliva, and an oral Yankar
suction machine can also be helpful; this device is similar to the suction
straw used in your mouth at the dentist oce. Ask your doctor or nurse
practitioner if medicines or a suction machine might help.
• Treat infections: Sometimes antibiotic medicines can help if your child
has a respiratory infection. If your child has a fever, a lot of coughing, or
greenish sputum (coughing up phlegm), he/she may have an infection.
A chest x-ray might be taken to help your doctor or nurse practitioner
diagnose an infection.
• Treat wheezing: If your child has wheezing due to constriction of the
bronchial tubes in the lungs, which can be similar to what a child with
asthma experiences, your doctor or nurse practitioner may prescribe a
nebulizer or inhaler medicines to open up the tubes that go to the lungs to
help your child breathe better.
Elevating the head of the bed or using extra
pillows can help with wheezing, as well as keeping smoke away from your child.
• Check for low blood counts: Hemoglobin carries oxygen around our
bodies. If your child has a low hemoglobin (Hgb) level, he/she may develop
shortness of breath or breathing trouble. If low blood counts are found to
be the cause of the breathing diculties, a blood transfusion can be given.
Seizures
Seizures are uncommon in children with DIPGs; however, sometimes they do
happen. Seizures can look dierent in each child. Some seizures are just mild
twitching or staring spells, while others involve shaking of an arm or leg or full
convulsions. Talk with your health care team and ask them if they think it is likely
your child might experience seizures. If you see any behaviors that are dierent
than normal in your child, ask your health care team if these behaviors might be
related to seizures. If your child has a seizure, the most important thing is to keep
him/her safe. DO NOT put anything in your child's mouth, such as a spoon or
your ngers. DO keep your child safe from things in the area that might hurt
him/her, for instance, move furniture away from your child and keep other people
(except medical help) away. It can be very scary to watch your child have a seizure,
but if you can remember, or ask someone else to remember, write down the time
the seizure started and stopped and what behaviors you saw. is will help your
health care team determine the best treatment for your child. Most seizures can
be controlled with medications.
Sleeping Problems
Certain medications can make it dicult for your child to sleep, especially
steroids. Anxiety and worry can also make it hard for your child to fall asleep
or stay asleep all night. Let your health care team know if your child is having
diculty sleeping. ings you can try to help your child sleep include:
• Having a regular bedtime routine every night.
• Starting quieter activities at least an hour before bedtime to wind down
an active child.
• Having your child take a warm bath or shower before bed.
• Wrapping your child in his/her favorite blanket or cuddling with him/her
before bed.
• Playing quiet, soothing music.
• Avoiding use of bright nightlights.
• Reading your child a story.
• Avoiding giving medications that may cause insomnia (sleeplessness) before
bedtime.
• Avoiding drinks or foods with caeine at least 2 to 3 hours before bedtime.
• Asking your doctor or nurse practitioner if a sleep medication might help.
Other Medical Issues
Your child may have medical devices, such as a central venous access device
Chapter 11: Caring for Your Child at Home
172
173
Chapter 11: Caring for Your Child at Home
(e.g., Broviac® catheter, Hickman® catheter, PICC line, Port-a-Cath®, Infuse-a-
Port®), a tracheostomy (a surgically created hole through the front of the neck
and into the windpipe), or a ventriculoperitoneal shunt (used to treat brain
swelling). Not every child with a DIPG has these types of medical devices, but
this section will give you some information about the use and care of some of
these devices. ese are only guidelines; you should follow the directions your
health care team gives you about how to care for any medical devices.
is section will also discuss complementary and alternative medicine (CAM).
Please discuss any CAM therapies that you might be considering with your
child’s doctor or nurse practitioner. Some CAM therapies interfere with
chemotherapy and radiation therapy,
so it is very important to let your health
care team know about any supplemental medicines you are using with your child.
Central venous access devices (central lines)
Central venous access devices are longer-term intravenous (IV) lines that can
be very useful for administering chemotherapy, blood product transfusions, IV
uids, total parenteral nutrition (TPN), and drawing blood. ese types of lines
are placed in large veins and the tip of the catheter ends in a large vein, just
above the heart. It is important to know that the catheter is NOT in the heart.
ere are several dierent types of central lines, and the decision about which
type of line is best will depend upon your child’s age, how frequently venous
access is needed, your child's activity level, and the ability of someone to care
for the line at home. e vast majority of children with a DIPG do not need a
central line, or if they do, it is usually for only a short period, such as needing
daily IV access for approximately 6 weeks with anesthesia during radiation
therapy for young children. Several dierent types of central lines are discussed
below. Your doctor or nurse/nurse practitioner is a great resource in helping you
decide if your child needs a central line, and if so, which type is best for your
child. e tips provided in this section are only general recommendations. You
should talk with your child’s nurse to nd out the specic care your hospital
recommends, who is responsible for providing the care, and who will teach you
how to care for the line at home.
Peripherally inserted central catheter (PICC line)
• Use: A PICC line is a more of a temporary line, which is often used for
shorter-term IV needs, such as with radiation therapy or a few weeks of
antibiotic therapy. It can be used to administer IV medications, blood
transfusions, and IV uids. A PICC line can also be used to draw blood.
• How it is placed: A PICC line is usually inserted under sedation or
anesthesia by an interventional radiologist or a surgeon. It is a small, exible
catheter that is usually inserted in the upper arm, with a couple of sutures
(stitches) to hold it in place, and then covered with an adhesive bandage
over the exit site. Part of the line will be external, coming out from the exit
site. e PICC line is usually loosely wrapped with an elastic bandage to
hold the line up out of the way until it is needed.
• Flushing the catheter: A PICC line requires daily ushing of the catheter
with a heparin ush solution, which keeps the blood inside the catheter
from clotting. e heparin ush solution is injected inside the catheter
and will not aect the ability of the blood in the rest of the body to clot.
• Dressing changes: e adhesive dressing over the exit site usually needs
to be changed once a week, or per your hospital's policy, or if the dressing
becomes soiled. Some health care teams recommend changing the dressing
less frequently, or only allowing a trained nurse to change the dressing, so
be sure to check your hospital's policy.
• Restrictions: It is recommended that children with PICC lines NOT swim
or submerge themselves in water (including bathwater). In general, contact
sports are discouraged. e PICC line is the easiest line to accidently pull
out, so you will need to take care to protect the line and teach your child
to protect it while playing or at school. If the PICC line is accidentally
pulled out, hold gauze or another clean bandage over the exit site until it
stops bleeding. Call your child’s doctor or nurse practitioner if the line
comes out and save the line so the doctor or nurse practitioner can be sure
all of the line was pulled out and none remains broken o inside the body.
If it is broken o, the catheter may need to be retrieved through a surgical
procedure.
• Home care: Your child’s nurse will teach you how to care for the PICC line
at home, including how to ush the catheter, how to change the dressing,
and how to administer medications through the PICC line (if needed).
e nurse will also arrange for the supplies that are needed to care for the
PICC line to be delivered to your home, and it is usually possible to have
a home care nurse come to your home for a few visits to help you care for
the PICC line until you are more comfortable doing it on your own.
Broviac® or Hickman® catheter
• Use: A Broviac® or Hickman® catheter is a longer-term central venous
Chapter 11: Caring for Your Child at Home
174
175
Chapter 11: Caring for Your Child at Home
access device. It can usually stay in from a few months to several years,
as long as it still works. A Broviac® or Hickman® catheter can be used to
administer IV medications, blood transfusions, and IV uids, as well as to
draw blood specimens.
• How it is placed: is type of catheter is usually inserted under sedation
or anesthesia by an interventional radiologist or a surgeon in an operating
room. It is a small exible catheter that is usually inserted in the upper
chest with a couple of sutures (stitches) to initially hold it in place; then
an adhesive bandage is placed over the exit site. Part of the catheter will be
external, coming out from the exit site. is type of catheter is also called
an external tunneled catheter, because the line has a cu made out of a
material called Dacron that helps your child's skin adhere to the catheter
and hold it in place. e surgeon makes an incision in the upper chest and
tunnels the catheter under the skin to the neck, where it is inserted into a
large vein. Generally, the sutures are not removed unless they are irritating
your child's skin or they fall out. Once the Dacron cu is anchored, usually
after about 1 to 2 weeks, it is dicult to accidentally pull it out. is type
of catheter can have one tube or be split into two or three tubes (called
lumens); each lumen requires daily care.
• Flushing the catheter: e catheter requires daily ushing of each lumen
of the catheter with a heparin ush solution, which keeps the blood inside
the catheter from clotting. e heparin ush solution is injected inside
the catheter and will not aect the ability of the blood in the rest of the
body to clot.
• Dressing changes: e adhesive dressing over the exit site needs to be
changed, usually once a week, or as per your hospital's policy, or if the
dressing becomes soiled. Some health care teams recommend changing
the dressing less frequently, or only allowing a trained nurse to change the
dressing, so be sure to check with your hospital's policy.
• Restrictions: Recommendations dier about swimming with a Broviac®
or Hickman® catheter. Check with your doctor or nurse/nurse practitioner
to see if your hospital recommends swimming with these types of central
catheters. Bathing is usually allowed, but the dressing needs to be changed
if it becomes wet. In general, contact sports are discouraged. If the Broviac®
or Hickman® catheter is accidentally pulled out, hold gauze or another clean
bandage over the exit site until it stops bleeding. Call your child’s doctor
or nurse practitioner if the line comes out and save the line so the doctor
or nurse practitioner can be sure all of the line was pulled out and none
remains broken o inside the body. If it is broken o, the catheter may
need to be retrieved through a surgical procedure.
• Home care: Your child’s nurse will teach you how to care for the Broviac® or
Hickman® line at home, including how to ush the catheter, how to change
the dressing, and how to administer medications through the catheter (if
needed). T
he nurse will arrange for supplies that are needed to care for the
Broviac® or Hickman® catheter to be delivered to your home, and it is usually
possible to have a home care nurse come to your home for a few visits to help
you care for the catheter until you are more comfortable doing it yourself.
PORT-A-CATH® or INFUSE-A-PORT®
• Use: A PORT-A-CATH® or INFUSE-A-PORT® (commonly referred to
as a port) is a longer-term central venous access device. It can usually stay
in for months to several years, as long as it still works. A port can be used
to administer IV medications, blood transfusions, and IV uids, as well as
to draw blood specimens.
• How it is placed: A port is usually inserted under sedation or anesthesia
by an interventional radiologist or a surgeon in the operating room. It is a
small, exible catheter with a port that is usually inserted in the upper chest
under the skin. Initially, one or two steristrips (buttery-type bandages) will
be placed over the areas where the port is inserted in the chest and where
the catheter is inserted into a large vein in the neck. is type of catheter
is also called an internal tunneled catheter, because the line is totally inside
the body. e surgeon makes an incision in the upper chest to put in the
port and tunnels the catheter under the skin to the neck where it is inserted
into a large vein. Generally, the steristrips are not removed and will fall o
by themselves with normal bathing. e port does not require the same
care at home as a PICC or Broviac®/Hickman® catheter.
• Flushing the catheter: Because the port is totally under the skin, it requires
a special kind of needle (a Huber needle) to reach the port for venous access.
In some cases, parents can be taught how to access the port, but in general
most of the port care will be done by a nurse. A port requires a heparin
ush once a month, but a ush may be needed more often if your child
receives medicines through the port. e port needle should be changed
once a week, if it is accessed.
• Dressing changes: No routine care is required for the port at home, unless
Chapter 11: Caring for Your Child at Home
176
177
Chapter 11: Caring for Your Child at Home
the port is accessed. If the port is accessed, the dressing should be changed
once a week with the changing of the port needle. Your nurse or home care
nurse will generally take care of the port.
• Restrictions: Unless the port is accessed, there are generally no restrictions
on swimming or bathing, but contact sports are discouraged.
• Home care: ere is usually no care to provide at home, unless the port
is accessed for medicines. If needed, home care nurses can come to your
home to teach you how to care for the port.
Complementary and Alternative Medicines (CAM)
Complementary and alternative medicines (CAM) are non-traditional
approaches to health care, but they are becoming more widely used in
conjunction with traditional medical care. Some CAM therapies can help your
child be more comfortable, but some can interfere with certain chemotherapies
or radiation therapies, so be sure to check with your health care team before
trying any CAM approaches. Make sure you go to a reputable CAM practitioner.
Check out the qualications and experience of CAM practitioners, just as you
would your doctor or nurse practitioner. Ask your health care team if they
recommend certain practitioners.
Some of these therapies may be provided in your
hospital by nurse practitioners and doctors who have specialized training in CAM.
Some CAM practitioners can come to your home. Examples of CAM include:
• Acupuncture
• Aromatherapy
• Art therapy
• Biofeedback
• Herbal therapies
• Hypnosis
• Massage
• Meditation and prayer
• Music therapy
• Pet therapy
• Play therapy
• Reexology
• Relaxation and guided imagery
• Reiki
• Tai Chi
• erapeutic touch
Practical Care Issues
ere are several other issues that may arise as you take your child home. Some
children have problems with balance and walking, communicating with others,
anxiety or emotional concerns, or central venous catheters or tracheostomy
tubes to care for. is section will discuss how to handle these challenges and
discuss other concerns for managing day-to-day life, such as traveling with your
child and practical tips.
Mobility
Some children with DIPG have ataxia (poor balance) or diculty walking due
to muscle weakness. Physical rehabilitation with physical and occupational
therapy may be very helpful to help strengthen your child, help your child learn
new ways to walk or be mobile with his/her physical challenges, and help you
to learn how to safely manage your child at home. Ask your doctor or nurse
practitioner to recommend a physical medicine and rehabilitation (PM&R)
doctor for an evaluation to help plan the best therapy approach for your child.
Inpatient rehabilitation services may be helpful, and outpatient or in home
services may be available.
Younger children can be carried, but it is helpful to have a good stroller for
longer distances. Remember that your child will grow, and the stroller you
had when he/she was an infant or small toddler may not be safe for him/her
as he/she grows. You will want to have a sturdy stroller to hold any additional
equipment or supplies your child might need. ere are several companies
that make heavy-duty strollers for medically fragile children. Ask your nurse
or social worker to help you nd the most appropriate stroller for your young
child. Some insurance companies will pay for this as a medical expense.
Older children may benet from having a wheelchair. Wheelchairs come in
many dierent sizes, so there is one that is the right size for your child. Ask
your doctor or nurse/nurse practitioner if they think a wheelchair might help
your child be able to move around better. Many parents feel a wheelchair is very
Chapter 11: Caring for Your Child at Home
178
179
Chapter 11: Caring for Your Child at Home
helpful because they can take their child outside and to places they like to visit.
Mobility concerns are not just related to walking or transportation from one
place to another. Your child may also have diculty standing or sitting, which
can impact daily activities such as taking a bath or shower, watching TV or
playing games, or even going to the bathroom. Discuss with your nurse/nurse
practitioner what strategies you can use to help your child manage his daily life.
Rehabilitation with physical and occupational therapy may improve strength
and balance so your child is able to do more things independently, but in the
meantime you may nd some assistive equipment particularly helpful. e
PM&R team may have even more suggestions, but some practical items to
consider are a shower or bath chair, rails on bathtubs or showers, walkers, canes
(there are many dierent types), hospital beds at home, and bedside toilets.
You
may want to consider moving your child's bed to a location in your house that
is easier for him/her to manage. For example, you can place the bed in a location
where your child does not need to go up or down stairs, or you can place it closer
to a bathroom.
Handicap parking permits
It may be possible for you to get temporary handicap parking tags or a placard
for your car to help you park closer to the hospital and other places you take
your child. is is particularly helpful if your child has impaired mobility. You
can obtain an application for handicap parking from your state Department
of Motor Vehicle Administration. Many applications are also available online.
ere is a section for you to ll out with the information about your car and
there is a section for your doctor or nurse practitioner to ll out to verify that
handicap parking permits are appropriate for you. Usually a temporary permit
is valid for 6 months but can easily be renewed. A placard is a good option if
you drive multiple cars, because it can be moved between cars.
Travel tips
Traveling with children can be challenging for any parent, but it may seem
overwhelming when you have an ill child. Travel is generally quite safe and your
doctor or nurse/nurse practitioner can help you nd ways to make things easier.
e following are some helpful tips that might help traveling go a bit smoother.
• Have the name, address, and phone number of a local hospital in the area
where you will be traveling, just in case your child needs medical attention.
• Ask your doctor or nurse practitioner to write a letter that summarizes
your child's history, the current treatment and medications your child
is taking, and how to contact him or her in case of an emergency. is
can be helpful in case you have to go to a local emergency room or visit a
doctor while you are traveling. If your child requires opiate medications
or injectable medications and you will need to carry syringes and needles,
ask your doctor or nurse practitioner to write a letter of medical necessity
for you to hand carry during air travel.
• If ying, take extra medications with you and pack at least a few days’ worth
in your carry-on luggage in case your luggage gets lost. If medication needs
to be refrigerated, remember to take cooler packs and notify ight attendants
that you need to refrigerate medications if it is a long trip.
• Call the airline, train, or bus company in advance and let them know
about your child's special needs. Ask for seats that have extra leg room.
Also let them know if your child has a wheelchair, walker, or other medical
equipment. When you arrive at the airport (or train or bus station), check
in early and again remind the attendants of any special needs your child has.
• If your child has other medication equipment (e.g., suction machine,
oxygen, feeding pump), ask your nurse or social worker if there is a way
to have this equipment supplied in the location where you are traveling so
you don’t have to carry all the extra equipment with you. Sometimes this
is not possible, so arrangements can be made to transport your equipment
with you as needed.
• Plan activities during the part of the day when your child usually feels best.
If your child needs to rest frequently, remember this and build that time
into your daily vacation plans.
• Plan your meals around your child's special dietary needs, if they have any.
• If possible, take an extra person with you to help with the logistics of travel.
• Keep things simple! Many family vacations create wonderful memories, but
not if everyone is stressed.
Build in time to relax and take things slowly so your
child does not become overly tired. Cranky children are no fun to travel with.
• Consider having friends and family travel to you. If travel is being planned
to visit relatives and friends it might be less cumbersome for your child and
family if visitors travel to you.
Communication Challenges
Many children with DIPG have trouble communicating at points during their
Chapter 11: Caring for Your Child at Home
180
181
Chapter 11: Caring for Your Child at Home
treatment. ese communication problems can stem from many causes such
as: swelling from the tumor, weak muscles in the neck that help the tongue
and mouth to form letters, or from the tumor irritating or damaging certain
cranial nerves that control the vocal cords. Some children with DIPG have
speech that is dicult to understand. Speech therapy may help. Children may
recover their speech over time if loss of speech is due to a reversible cause, such
as swelling. Sometimes speech does not improve much, even with therapy. is
can be quite frustrating for you and your child. Remember to be patient and
let your child have extra time to try to communicate with you. Here are some
helpful hints to improve communication.
• Speech therapy: Ask your doctor or nurse practitioner if he or she thinks
speech therapy would be helpful and, if so, to help you arrange the therapy.
• Picture boards: Your speech therapist can help you make a picture board
to identify common things that your child may be trying to communicate.
You can also do this as a family project. Find pictures in magazines, print
pictures from online, or draw pictures of common things your child may
want to communicate, such as being hungry/wanting food, being thirsty/
wanting a drink, wanting to go to bed, wanting to go to the bathroom,
wanting to play, wanting to watch TV or a DVD movie, having pain,
feeling nauseated, etc. Some speech therapists have picture boards that are
already made for dierent ages of children. You can teach your child to point
to the picture of what he/she is trying to communicate. ere is an iPad
application (app) called Picture Board that is available for free download
from the Apple store.
• Voice communication devices: ere are computer programs or hand-held
electronic devices that allow your child to push a picture of what they want
and the computer "speaks" the word or phrase. ese may not be available
in your hospital, but you can ask about ways to order them. ere is a useful
iPad application (app) that can be downloaded for free called “Talk Assist”
for those children who can type text that is then converted to speech; and
another free application called “Small Talk, Conversational Phrases,” which
provides a vocabulary of pictures that subsequently talk in a human voice.
• Pencil and paper: Have pencils (or markers or crayons) and paper readily
available for your child to write down what he/she is trying to communicate.
Save the common cards so they can be used over again. If your child cannot
write but can read, consider writing common phrases out on 5 x 8 cards for
your child to use to communicate what he/she needs or wants.
• Take your time and allow your child to take their time: Diculty
communicating is very frustrating for most children. Try not to rush your
child or ask him/her over and over to repeat what he/she is trying to say.
Begin to use picture boards or the written cards early, when your child is
not having much diculty communicating, so that it is natural to him/
her when he/she is no longer able to speak clearly.
Chapter 12 in this book provides additional information on the communication
needs of the child diagnosed with a DIPG.
Other Helpful Tips
Other challenges may arise that are not anticipated. Every child and every
family is special and may have special needs. Ask your health care team about
any issues that have not been mentioned here. In addition, here are a few things
to consider.
• Clothing: Your child may gain weight or lose weight as a result of
treatments. Some children develop sensitive skin, where some fabrics are
irritating. Have a supply of loose-tting clothing that is easy to get on and
easy to remove. If your child has muscle weakness or is not able to stand,
he/she may not be able to dress himself/herself like he/she used to. Clothing
with elastic around the neck and leg openings is particularly helpful. You
may also need to have several sizes of clothing available for times when
your child gains or loses weight. Buying new clothing can be expensive,
especially when your child may not be the same size for any length of time.
Consider visiting second-hand or discount clothing stores to get basic items
and save more expensive clothing purchases for special occasions. If family
and friends ask how they can help, consider asking them to provide gift
cards for clothing stores that your child might like.
• Financial worries: e costs of treatment and medical equipment can
be substantial, even if you have great insurance coverage. Most insurance
companies do not pay for everything your child will need. Anticipate out-
of-pocket costs and try to budget for them, if possible. Ask your social
worker if your child might qualify for Supplemental Social Security Income
(SSI) or other possible sources of additional funding to help with some of
the costs. Consider letting family and friends who are asking what they
can do for you, to help with fundraising events to raise money for these
unanticipated medical and personal expenses. In addition, many hospitals
charge for parking and meals in the hospital’s cafeteria and these can add
Chapter 11: Caring for Your Child at Home
182
183
Chapter 11: Caring for Your Child at Home
up after a while. Extra funds can come in handy for these types of expenses,
which are usually not covered by insurance.
• Durable medical equipment: Equipment that your child needs is called
durable medical equipment (DME). It is important to know this term,
because this is how benets for payment of equipment are determined by
your insurance company. Your nurse should review any equipment that your
child might need at home, but most equipment is provided by an outside
company that your insurance company recommends. e equipment that is
used in the hospital may dier slightly from what is delivered to your home.
Make sure you are given instructions about how to use the equipment,
how to troubleshoot common problems, who to call 24 hours a day in an
emergency, and who to call when you have routine questions. You should
also ask to have a home care nurse come for a few visits to help you learn
how to use any DME. If the equipment company does not provide a set of
written instructions for each piece of equipment, ask for them. Keep this
information in a binder or notebook in a place that is easily accessible to
anyone who might be caring for your child. Be sure to put the emergency
phone numbers for each company that provides DME in the beginning of
the notebook or in a place that can be found quickly, if needed.
Making Dicult Decisions
Your child has a rare and challenging tumor and many dicult decisions will
have to be made along the way. It is helpful to have conversations about the
dierent types of situations that may come up before they actually do; that way
your family and your child (if he/she is old enough to help make decisions about
his/her health) will be able to talk about these situations openly and honestly.
Some of these decisions may be dicult to talk about, so talking about them
early, when there is no urgency, can make it easier if the need arises to make
urgent decisions. Your social worker will be a wonderful resource to help you,
your family, and your child talk together about dicult decisions. ese are
some helpful tips about making dicult decisions.
• Honesty: Have open discussions with your family and your child (if he/
she is old enough to understand and participate in decisions about his/her
health) about your child's diagnosis of DIPG and what the prognosis is,
according to your doctor or nurse practitioner. Most children are thinking
about these things, even if they are not talking about them. Sometimes
your child's imagination may be worse than reality, so talking about what
may or may not be happening will actually help him/her worry less. Be
honest and do not lie to your child. Children are usually very good about
guring out when parents are lying to them.
• Hope for the best and prepare for the worst: Your health care team is
working very hard to treat the DIPG and help your child. ey may or may
not have given you statistics about cure rates and/or relapse rates for DIPG.
However, statistics really do not mean a thing, because for your one child,
there will either be a 100% cure or not. Try not to let statistics keep you
from enjoying life in the moment. Enjoy every day with your child. Keep
your hopes up that your child will be that 100%, but talk with your health
care team about what to expect if your child does not survive the DIPG.
• Five Wishes™ or My Wishes™ or Go Wish™ Cards: Some children and
families nd it hard to talk about making dicult decisions and/or facing
end-of-life care decisions. Be sure to talk with your social worker about
your concerns.You may also nd it helpful to talk with a counselor or
psychologist. Several other tools can also help you with these conversations,
such as Five Wishes™ and My Wishes™, which are decision-making tools.
Five Wishes™ is a set of questions for older children, adolescents, and
young adults, and My Wishes™ is better suited for younger children
or developmentally delayed children. ese tools have several dierent
questions about who your child might want to make decisions for him/her
if he/she is not able to, and other health care decisions he/she might want
people to know about. Go Wish™ Cards are like playing cards that have
phrases about health care decisions written on them. Ask your health care
team if you can have a copy of these tools to use at home. Your local hospital
might use other tools that are equally as good as the ones mentioned here.
• Other books and pamphlets: Several wonderful books are available to
help you with making dicult decisions. Ask your health care team to
recommend some of these to you. ere is also a list of books in the appendix
at the back of this book that you might nd helpful as well.
While the journey through DIPG treatment can be physically and emotionally
challenging for both you and your child, the advice listed in this chapter can
help you feel more prepared to care for your child at home. Remember that
your health care team is there to support you through this dicult time, as
are parent support groups led by others who have walked the same path your
family is now walking. Reach out and ask for the help you need to best care
for your child with DIPG.
Chapter 11: Caring for Your Child at Home
184
185
Chapter 11: Caring for Your Child at Home
Parent Perspectives
I didn't understand all that would be involved in Andrew's care at home until
we left the hospital. There were medications to dispense. He could not be
left alone for extended periods of time as he was very unstable. If he was
in his hospital bed or strapped into his wheelchair, we could leave him for
a short time—as long as we were close enough to hear him if he needed
us. He could not walk or get down on the floor without our help. He also
needed help with bathing and toileting. In the hospital we had people to
help with all of these different things. At home we were on our own. This
did not change how we felt about having him home; it just made being home
very different from what that meant before he was diagnosed with DIPG.
Stella eats very little and requires liquids to be thickened to enable her
to swallow them. Her diet at the moment consists of avocados, mashed
potatoes, apple sauce, hamburgers, ice cream and milk. Since October
she has been incapable of sitting up unassisted, and as of December she
is unable to hold her head up without support. She drools constantly and
has begun to suffer from seizures.
When our son was diagnosed with DIPG, each of our lives were impacted
in some way. While he bore the physical effects of the tumor itself, each of
us were changed because of it. As we lost the child we knew to steroids—
not once, but twice—we grew to understand that the real boy we knew was
not the body we saw with our eyes, but the soul we loved with our hearts.
I am a single parent and had to care for Warren alone. Warren’s dad would
take him on the weekends so that gave me a much needed break. I also had
family and friends that would watch Warren when I needed to go to the
store or something, but Warren didn’t want me to leave him so I naturally
tried not to.
I took a leave from work after Warren was diagnosed. I live in income-based
housing so that worked in my favor. Even with all that, I had no way to pay
other bills and pay for gas to get us back and forth to the doctor and such.
Then our community stepped in and helped out with fundraisers and such.
The people around where I live were so wonderful and caring and because
of them I didn’t have to worry about paying the bills.
From the beginning, I believed that complimentary care was an integral
aspect to Alexis’ overall treatment, and medical plan. In our initial
discussions with Alexis’ treatment team, it was important to discuss
complimentary care. From those first few moments we landed headlong
in the childhood cancer community, we were given the proverbial “green-
light” to use supplements as long as we cleared them with the team first.
Initially, we had a minor debate regarding the use of antioxidants. The
conventional wisdom of the past suggested that the use of antioxidants
could negatively impact the efficacy of radiation. After doing research on
the topic, I was able to demonstrate that this in fact was nothing more than
anecdotal information. We quickly began consulting with a nutritionist that
many other pediatric and adult brain tumor patients worked with. Whether
the use of supplements and the complimentary care had any impact upon
Alexis’ overall course we ultimately will never know of course. I certainly
would like to think that it did. For around twenty months of Alexis’ battle,
from sun-up to sun-down, Alexis willingly took approximately twenty
different natural supplements and vitamins. I tasted everything prior to
administering them to her. Some were simply awful so we devised strategies
for getting them into her. In the end, Alexis took everything like a trooper.
It seems to be taking more effort for our son to chew and to form words. So
he is not chewing things as well as he was last week, and he is choosing not
to use his words as much as he normally does. (I need to throw in here though
that he uses his words quite loudly and clearly when he wants me and I'm
not responding quickly enough!) He has been vomiting periodically—almost
always later in the day. This has become more of a problem over the past
week, so there has been some concern regarding nutrition. The vomiting
is believed to be a gastrointestinal issue rather than a neurological issue,
and a scope this afternoon did show delayed gastric emptying. We have
made some medication changes and some food changes, and have begun
using an NG tube for supplemental nutrition.
Chapter 11: Caring for Your Child at Home
186
187
Chapter 11: Caring for Your Child at Home
AmbuCab arrives about 7:15 a.m. on dressing change mornings. By the time
we load and unload, we end up at the hospital by 8:00 a.m. The AmbuCab
driver pushes my son to the fourth floor while I push another wheelchair
piled high with our stuff (dressing change supplies; his Bi-PAP; a backpack
filled with a change of clothes, medications, and other necessary things;
my purse, and my laptop). We arrive and I begin to arrange the room in
preparation for the big event. Shortly before 9:00 a.m. I set up the Bi-PAP
for use while my son is under sedation. A nurse or tech prepares him by
attaching electrodes to his chest to monitor vital signs. The wound team
arrives to open dressing change supplies, and the intensivist arrives to
administer propofol for sedation. By 9:00 a.m. the tiny room is full with at
least five people, and the dressing change begins. Someone from phlebotomy
arrives just after 9:00 a.m. to draw blood, and someone from the I.V. team
still comes on Mondays to change the needle in the port. We have noticed
that the dressing changes are getting shorter and that we are using fewer
supplies. The few wounds that remain are clean and steadily improving,
and my son now rarely experiences pain.
Caleb was not able to eat and I was worried he would be hungry. He hated
being hungry. So, they put in an NG tube and kept food going into his tummy
to make sure he would not ever feel hungry. He was restless at times and we
had valium on request. If he indicated pain, we had morphine on request.
He was still able to get up (with assistance) to use the bathroom, sit in a
chair, watch TV. He could not talk very well but we developed methods of
communication during those days—first with hand signals, then with eye
movements.
To help control her eating we implemented the use of a timer and a second
hand clock that would show time running down using color. When either
thing beeped Peyton knew it was snack time. We would get sneaky once in
a while and add more time to the timer when she wasn't looking. We also
provided little snacks and made sure we had an activity planned for after
snack so she would be focused on something else. Sometimes I made stuff
up like, "Oh we are out of butter to make cookies." Distractions help.
On a side note one of the best things we did during our daughter's illness
was strabismus surgery. It fixed her eyes, hence eliminating the need
for blacking out one lens of her glasses. Most importantly, it gave her
confidence, made her feel better, and gave her a sense of accomplishment.
We are settling into a routine at home—though we are still in limbo in some
ways. Our living room looks like a Pediatric Intensive Care Unit, with the
focal point being our son's hospital bed. We've done our best to organize
so that the medical equipment and supplies are easily accessible, but not
obvious to the casual observer. We are set up to use oxygen; to administer
I.V. fluids and medications; to take care of wound care and our son’s port;
to monitor oxygen saturation, blood pressure, heart rate and temperature;
to use Bi-PAP and suction. We find ourselves using Bi-PAP and oxygen only
when he sleeps; he has not required suctioning since he had a cold in early
July. We do administer I.V. medications and fluids daily as there is question
regarding his ability to absorb oral medications and fluids.
We met with the neurosurgeon, who explained that surgery was not possible,
nor was a biopsy because of the location. She answered questions that we
had written down, and ultimately told us that other children who had this
tumor did not survive. When we asked how long Bryce had, we were told
that with treatment, these children typically had “more or less than a year.”
I remember looking at my husband, and saying that one year was going to
go too fast. And I remember looking at the doctor and doing what I now
call the most important thing that helped us to live through this. I asked
her to hook us up with anyone and everyone who could support us through
this—psychologists, social workers, doctors, whomever. And she did. We
thank her every day for that.
We were told to talk about everything that was important, and about
what was coming, so that we could be sure that we had those important
conversations before speech became an issue, as it typically did with DIPG.
That’s not to say that we didn’t continue to research, to check out a trial
offered in Toronto, or search the world over for a cure. But we also decided
Chapter 11: Caring for Your Child at Home
188
189
Chapter 11: Caring for Your Child at Home
at that point that if the treatment available would not allow Bryce to LIVE
the way he chose to every day, then we would respect that. He was 13, and
at that point he had already voiced that he did not want surgery, or to have
chemo because he was afraid of having a port put in. So because nothing
was curative, we did not have to try to change his mind.
We went back home. The thing that became obvious over the next few weeks
was that Bryce just wanted to go back to school, to a regular routine, and
to be a regular kid. He didn’t want to be in the spotlight, babied, coddled,
or have all of the attention that others wanted to give him. He even went
on his Grade 8 graduation trip. And not until months later, did he know
that we were also in Cleveland that day, just in case something happened
and he needed to be rushed home. Thankfully, he never needed to know.
Bryce’s progression surprised everyone because the doctors thought that
if the onset of new symptoms were swift, that Bryce would only survive a
very short time. He, however, actually survived for two months after his
progression date. Over that time, he also developed pain—neuropathic
pain—in his knees, and feet. We tried everything—massage, ice, heat,
arnica cream, A535, Reiki, Dilaudid. Nothing gave him much relief. And
we had to step up and become his advocates with everyone—medical staff
and visitors to make sure that his wishes were met. That was no small feat.
At first eating wasn’t much of an issue. For most of the time Warren could
feed himself, but would need a little assistance. As he started to struggle
I had him eat softer foods like mashed potatoes and I pureed things in the
blender. His biggest problem was he couldn’t get his mouth open very wide so
getting food in was difficult. I had to buy smaller spoons and deeper spoons
so he could get a decent bite. He also could only drink using a straw. The
straw had to be placed in the right side of his lips or he couldn’t get a drink.
Meal time took about an hour. Warren would eat in his "stander"—a big
wooden device that I strapped him into. He was in a standing position
and there was a tray in front for games and food and such. Sometimes he
would sit on the floor but he couldn’t hold himself up so I had to prop him
up against the couch and slide our coffee table over him so he was stuck
in between. I then had to put pillows on either side of him to keep him from
falling over. Once Warren started taking steroids he wanted to eat more
often, so rapidly began growing out of his clothes. I had to buy all new
pants (with elastic bands), shirts, underwear and jammies.
We bathe Andrew and dress him daily. We move him—by Hoyer lift—from
bed to wheelchair and from wheelchair to bed several times on any given
day. We do our best to keep his hands busy because the more he uses them,
the better. He enjoys shooting various Nerf guns at targets we hang from
his Hoyer lift. (He also enjoys "shooting" at his brother!) Andrew has
physical therapy at home three times each week. We do our best to guard
that precious time with his therapists and to work with him ourselves—-as
time allows—on other days.
Long shirts that look like dresses were awesome for our daughter. Paired
with stretchy leggings; it was a good thing. All the name brand stores had a
wonderful selection of different colored leggings. We didn't want Peyton to
feel constricted in her clothes, so I usually bought the next size up as well.
I was stunned. Sick. I was left alone in a room in the PICU to take care
of my very sick daughter while also trying to accept the fact that I would
have to watch her die. My brain tried to reject the concept like my stomach
would reject tainted food. At that point, I shut down. I let the doctors know
that they should only talk to Joe, not to me. I could not handle it. I was
struggling to keep myself from letting the panic and disbelief take over. I
couldn’t eat. I couldn’t sleep. I didn’t know what to do with myself. One
thing I did know. No matter what I was feeling, I had to get over it and be
there for Bizzie. So I did…somehow.
Our families reacted much how we suspected they would—with tears of
course, but with tremendous support and strength as well. We are certain
many other emotions were felt privately but in our presence and in the
presence of our children there was nothing but complete strength displayed.
Everyone spoke honestly about Liam's illness; however in those early days
we made a decision that we would not tell our kids or Liam about the course
this disease generally takes. Their little hearts were already burdened with
so much. As Liam's condition improved or declined we had honest age-
appropriate discussions at every step. Our kids were a tremendous help to us
Chapter 11: Caring for Your Child at Home
190
191
Chapter 11: Caring for Your Child at Home
and to Liam. We included them as much as we could in his care. Our oldest
child, then in third grade, would read to him; his twin brother "assisted"
me with dressing changes and his little brother, only three, came to every
day of radiation and every clinic visit. But mostly and most importantly
they continued to play and treat Liam as they always had. Liam loved this.
He did not want to be different. It was a wonderful gift they gave to him.
On April 1st, my son was admitted into hospice care. With my nursing
background, hospice acted in his case more as a sounding board for us
as we did all of his care ourselves. In crisis moments they helped with
medication calculations, gave suggestions on meds that might help ease
his pain and they offered a wide range of therapies. He enjoyed Reiki and
therapeutic touch. He received weekly visits from therapy dogs. This was
such a blessing for a little boy who had a strong and special connection with
animals. We had artists who are friends and teachers to our other children
come and do art projects with our kids and at one point near the end of our
son’s journey a friend came with his guitar, invited all the neighborhood
kids to come and they sang songs together. It was a beautiful afternoon.
Brendan welcomed the idea to have an NG tube, then G-tube then "Micky"
button so we could deliver meds and nutrition easily. He felt better when
he could get meds without choking and nutrition without having to chew
and swallow. Keeping him hydrated and fed was key to keeping him strong
and enjoying a good quality of life especially when the chemo caused GI
issues and he lost his appetite.
A few weeks later, Aimee woke with a severe headache, and was partially
paralyzed on the right side. When I called her new doctors, they put her
back on the dexamethasone. I also began her on a drink I heard about from
another mother whose child also had a brain tumor. The drink was called
Vemma, which is made from the Mangosteen fruit. Once she began taking
the Vemma she regained full strength on her right side.
Aimee was doing well since she began the Vemma, so she demanded to
return to school in September. She was entering the 7th grade in a brand
new school in a new state and knew no one in her class. I personally did
not want her to attend but she felt it was something she needed to do. A
few days after school started I found a letter that she had written to her
classmates.
"Hi, I am Aimee. I am 12 years old, and I am just like you. I love reading,
music, go-kart racing, cheerleading and making crafts. Yes, I may be
sitting in a wheelchair, and my face may look funny and talk funny, but
please don’t be afraid of me because you cannot catch what I have. I have
a brain tumor and the doctors say I am going to die. But I just want to be
as normal as possible, just like you. So please don’t be afraid of me. I am
not afraid of you even when you make fun of me. I will still be your friend,
so can you please be mine."
We were very busy. My son had his radiation done an hour and half from
our house. So while he was doing radiation we would get up about 7:30
or 8:00 a.m., have breakfast, go to physical therapy and/or occupational
therapy for 1 to 2 hours. We'd then go home and have lunch and rest and
play, then leave about 3:00 p.m. for a 5:00 p.m. or 6:00 p.m. radiation
appointment. We would be there for 30 minutes or so then would drive back
home. We would get home about 7:00 p.m. or 8:00 p.m., eat if we hadn’t
already, and take a bath when needed or felt like it. Then we would go to
bed because Warren would be tired.
If we had a doctor’s appointment then we would skip physiotherapy and
occupational therapy and drive the 3 ½ hours to the doctor, then stop on
the way back home to have radiation if it was scheduled.
My son didn’t go back to school after diagnosis because we were too busy
with radiation and such and after that I just wanted him home. He couldn’t
write or walk and had a hard time speaking and would have had to have
constant care with feeding and going to the bathroom.
By April he began to require the use of narcotics to manage pain as a result
of steroid-induced skin breakdown. Just before he was transferred to PICU
on April 9th, he was placed on a continuous drip. As we began to prepare to
go home in May, the importance of oral (as opposed to I.V.) medications was
discussed, and he was slowly switched from Fentanyl (by I.V.) to Methadone
(by mouth). He was on such a small amount of Methadone that we began
Chapter 11: Caring for Your Child at Home
192
to wean him off the medication completely last weekend. We noticed that
he did not seem well, but it was not until Tuesday that we began to realize
it was the lack of Methadone that was causing him to feel miserable.
We discussed the way our son was feeling with his doctor that afternoon, and
she made the decision to put him back on Methadone. When he received a
bit of fast-acting morphine to help him until the Methadone had opportunity
to take effect, it was almost immediately as if we had flipped a switch. He
went suddenly from misery to contentment. One moment he was telling us
repeatedly that he could not get comfortable; the next moment he turned
into a chatterbox...with a smile on his face.

193
Chapter 12: When a Child Can No Longer Speak
Chapter 12
Communication: When
a Child Can No Longer
Speak
David Brownstone, MSW, RSW
Caelyn Kaise, MHSc, SLP(C), Reg.CASLPO
Ceilidh Eaton Russell, CCLS, MSc (candidate)
Supporting a child or teenager who has a brain tumor is an incredibly important
and dicult job. And trying to help them understand and live with their changing
abilities can be overwhelming, especially when caregivers naturally struggle with
these changes themselves. e situation is also a challenge because while a child’s
physical abilities to communicate—including the ability to produce speech and
to express thoughts and feelings—can change, cognitive abilities often stay intact.
So if a child or teenager has trouble communicating because of a brain tumor,
the task of supporting them becomes even more complex.
Family members and caregivers who have been in this situation often express that
they did not know what to do or where to start, and they often felt helpless and
frustrated. But in the end they did it. With time, patience, creativity, and support,
families nd ways to communicate with their children and teenagers with brain
tumors, even though these young patients had, or have, trouble speaking.
is chapter includes “lessons learned” from talking with 14 families about their
experiences, as well as our team’s experiences working with families of children
with brain tumors. (Note: Our team only
interviewed families of patients younger than
age 13 and our examples reect this. While
the examples may not be relevant to teens, as
many issues and struggles are unique to that
age group, the communication strategies are
similar and can be adapted for teens.)
David Brownstone is a Social
Worker with the Brain Tumor
Program and an Academic
and Clinical Specialist in the
Department of Social Work at
the Hospital for Sick Children in
Toronto, Canada.

Chapter 12: When a Child Can No Longer Speak
194
195
Chapter 12: When a Child Can No Longer Speak
e parents we spoke with generously shared the creative strategies and tools they
developed, the most important conversations they had, and the most important
lessons they learned. When we began to talk with these parents, our goal was to
develop a new communication tool, yet they taught us that although tools are
helpful, in the end, direct communication is more valued and helpful. Families
encouraged us to share strategies and resources with families like yours so that
others facing this stage of brain tumors will have ideas about where to start, what
to try, and know that they—and you—are not alone.
Of course every family and every child is unique, each with their own values,
philosophies, experiences, and backgrounds. Some of your family’s experiences
may be very dierent from those of the families we interviewed. However, some
of the situations they faced or the strategies they tried may be similar to yours
or helpful to you. We encourage you to think about the ideas outlined in this
chapter and to use or modify them so they work for your family and are well
suited to your child’s age and developmental stage.
Most of all, the families we talked to and the members of our team sincerely hope
that sharing this information will help you and your family feel, at the very least,
a little more prepared and supported during this dicult time.
Quick Tips
To help you focus on how to approach and enhance communication with your
child, here are a few quick tips to think about.
• Practice communication strategies before your child needs to use them.
• Practice more than one signal for “yes;” no response can be used to mean
“no.”
• Start by asking broad questions, and then ask more and more specic questions
as you get an idea as to what your child is thinking about or wanting to say.
• Use simple sentences to get to the main point. For example, ask “Are you
hungry?” instead of “Do you want something for dinner?” Remember the “KIS”
principle: “Keep It Simple.”
• Remind your child what the “yes” signal is before asking each question.
• Wait longer than usual for your child to
respond.
• If your child has a hard time responding,
repeat the question or simplify it. For
example, if you’ve asked “Are you hungry?” simplify by saying, “Hungry?”
• To make sure your child’s message is understood correctly, repeat what you think
he said. For example, “Okay, you are hungry,” or “So you’re not hungry.” is
gives your child a chance to conrm that his message was interpreted correctly.
• Be patient with yourself, your child, and the process.
• When exploring emotional issues, ensure that you understand your child’s
unique perspective rather than thinking about it only from an adult perspective.
In other words, focus on how your child is thinking and feeling, not how you
would think or feel in the same situation.
Communication
Preparing for the unexpected
It is hard to prepare for something when you don’t know what to expect. Brain
tumors aect children’s abilities in dierent ways at dierent times, but some
changes are more common than others and can be anticipated. For example,
speech often starts sounding slurred and can be dicult to understand due
to weakness or diculty coordinating the lips, tongue, and jaw. Children’s
abilities to use their arms and hands may also become compromised, making
it dicult for them to write, draw, or point.
Regardless of the kinds of diculties children with diuse intrinsic pontine
gliomas (DIPGs) have, the parents we interviewed agreed that two important
strategies helped maximize communication with them.
1. Learn and practice ways your child can communicate without speech
before your child needs to use them. is is not always easy. Children
can be reluctant to use communication strategies before they absolutely
have to, and parents and children often do not want to think about a
time when these strategies will be necessary. is reticence is natural
and understandable. However, the patience and concentration that are
needed to learn a new skill may not be present once your child’s energy
and abilities are declining.
2. Practice more than one way of
communicating without words. is
way, if some of your child’s abilities
change in an unexpected way, she
can continue to communicate using
Caelyn Kaise is a Speech-
language Pathologist with the
Brain Tumor Program at the
Hospital for Sick Children in
Toronto, Canada.
Ceilidh Eaton Russell is a Child
Life Specialist with the Max and
Beatrice Wolfe Children’s Centre
at the Temmy Latner Centre for
Palliative Care, and a Researcher
at the Hospital for Sick Children in
Toronto, Canada.
Chapter 12: When a Child Can No Longer Speak
196
197
Chapter 12: When a Child Can No Longer Speak
another familiar means. When practicing other ways to communicate,
it is often useful to nd a way to adapt a current communication tool or
technique to suit your child’s changing abilities rather than switching to
a brand new system. By adapting a strategy that children and families are
more familiar with, their experiences serve as “practice” and they may feel
more comfortable and condent in their abilities to use it.
In this chapter, we present some concrete examples of communication tools
and strategies to use with children who have a DIPG. is is in no way an
exhaustive list, but it serves as a stepping stone to understand how to maximize
communication.
Dierent ways to ask questions
Two techniques that are very useful when helping children express a wide
range of messages are:
1. Oering two clear choices.
2. Asking questions that can be answered with a “yes” or “no.”
ese techniques require you to ask clear and carefully worded questions and
will take thought and practice.
Oering two clear choices
No matter what a child’s functional ability is, he/she is likely to be able to
choose between two things, whether by pointing at or by looking at dierent
objects. It is important to clearly tell your child what the two choices are and
then ask your child to show you which one he wants.
For example: A parent can hold chocolate milk in one hand and juice in the
other. After showing them to the child and saying what is in each hand, the
parent then asks the child which one he/she would like, reminding him/her
to point or look at the drink he/she wants. Once the child has made a choice,
the parent should double-check by asking, “Do you mean that you want the
juice?” then wait for him/her to show that he/she means “Yes.”
When a child is choosing between two things that you cannot show him/her,
try asking a series of questions to nd out what he/she wants. For example:
1. “I wonder if you would rather go for a walk or take a bath?”
2. “I’ll ask you about one thing at a time, and then I’ll wait after each one
in case you want to say “yes.”
3. “So, want to go for a walk?” After asking this question, pause for at least
10 seconds.
4. If your child does not respond, say, “Okay. Want to take a bath?”
It may take your child longer than usual to make a choice, so remember to
wait for a response. If your child does not respond, here are a few things to try.
1. Ask if he/she needs you to remind him/her of the signal for “yes.”
2. Ask if he/she needs you to remind him/her, what the options are, then
wait for him/her to respond. If he/she says “yes,” repeat the series of
questions above and wait for his/her response.
3. Ask if he/she does not want either of the choices that were oered, and
wait for him/her to respond. If he/she says “yes,” try to think of other
options he/she may prefer.
Oering choices helps children feel like they have some control. Although
with this method it can take a long time to nd out what your child wants,
it is usually worth the extra eort.
Using “Yes” or “No” questions
Even when it’s very dicult for children to choose between two things,
caregivers can help them express themselves by asking questions that can be
answered with a simple “yes” or “no.” is is a technique that can be used
with a wide range of other communication tools and techniques and a method
that will come up numerous times throughout this chapter.
Children can show they mean “yes” in a range of ways, including:
• Nodding their heads.
• Giving a “thumbs up.”
• Wiggling a nger up and down.
• Raising their eyebrows.
• Looking up (like nodding with their eyes).
• Wrinkling their nose.
• Wiggling their toes or moving a foot.
**Remember to practice more than one signal for “yes!”
Chapter 12: When a Child Can No Longer Speak
198
199
Chapter 12: When a Child Can No Longer Speak
Any part of the body that the child can control can be used as a signal for
“yes.” Instead of making a second signal for “no,” it is easier to assume
that if the child doesn’t say “yes,” he means “no.” is creates less confusion
about which signal to use for which word. When practicing this technique,
ask your child to choose a couple of signals, then ask him/her ve “yes” or
“no” questions that you know the answers to and make sure your answers
match his/her signals. If they match, you’re ready to start!
Some questions may have more than one meaning, so it is very important to
ask in a way that is clear and direct. For example, asking your child, “How are
you feeling?” can be confusing, because it could refer to physical or emotional
“feelings.” Instead be specic; ask “Are you sad?” or “Does your body feel
okay?” is allows your child to respond with a clear “yes” or “no.”
When there are fewer clues about what your child wants or needs, start
by asking broad questions, then ask more and more specic questions
based on your child’s responses. For example, if your child seemed upset you
could start by asking, “Is something bothering you?” If the answer is “yes,”
you can ask more specic questions one at a time until she says “yes” again.
e following is an example of a progressive series of questions.
1. “Is it something in your body that’s bothering you?”
•Ifshe says “yes,” ask, “Isityourhead?”or “Isit yourstomach?”
continuing to ask about dierent body parts until she says “yes.”
Remember to pause after each question to wait for a response.
o Once your child says “yes” about a particular body part, ask,
“Is it sore?” “Is it itchy?” or “Is it hot?” etc.
If he/she does not say “yes” to any part of the body, say “Okay, maybe it’s not
something in your body that’s bothering you. Are you feeling upset about
something?” If he/she says, “yes,” ask questions about specic feelings, such as,
“Are you sad? Are you feeling frustrated?” until he/she says “yes” to something.
If your child does not say “yes,” try asking, “Is it something you’re thinking
about?” or “Are you worried about something?”
is example illustrates that it is often easier to know what to ask when the
topic is concrete, such as physical sensations or nding out what a child
wants to do. Talking about more abstract concepts, such as emotions and
ideas can be much more complicated because there are many more possible
questions. Because of this, you will need to ask a lot more questions when
discussing these topics.
If you continue to ask questions without being able to gure out what your
child wants and he/she becomes frustrated, it is good to talk with your child
and explain in the following way.
1. “I know that you know what you want to say. is is really hard for both
of us, but I want to try to help.”
• enask,“ShouldIkeeptryingtogureoutwhatyou’rethinking
or should we take a break? I’m going to ask you that again and wait
for you to show me “yes” after the one that you want me to do.”
o en repeat these two options, pausing in between for your
child’s response.
It is especially important to be patient with the process, and with yourself
and your child, during these discussions.
Figuring out what to ask
Although interactions may feel dierent when a child has trouble speaking,
he/she is still the same person as before. Try to consider past experiences with
your child, including his/her typical behaviors, preferences and needs, to give
you clues about what he/she would want now.
Facial expressions and body language
When you recognize a familiar facial expression, it probably means the same
thing it used to mean. In addition to telling you about their feelings and
moods, a child’s face or body can also show you whether he/she is comfortable
(through a relaxed body) or uncomfortable (through a tense body or face).
Tumors may aect facial muscles for some children, making facial expressions
look dierent than they used to. However, parents often say that even with
these changes they can recognize what their child is expressing, especially
because the children’s eyes continue to show a lot of emotions.
Routines and preferences
Time of day, familiar routines and the context of a situation can oer clues
about whether your child is tired, hungry, wants to bathe, go outside, or
play. Although children may have to do these things in a dierent way than
they used to, if they are losing some of their abilities it is still helpful and
comforting for them to participate in familiar activities as frequently as
possible. inking about the situation—where you are, who’s around, and
what you are doing—will also help narrow down the questions or the needs
Chapter 12: When a Child Can No Longer Speak
200
201
Chapter 12: When a Child Can No Longer Speak
the child may currently have.
We have found that while it may feel like there are a million things a child
could want or need; it is often the simplest things that the child wants. Try
to always start with basic questions, such as whether the child wants to sit
up or change position. If the child has a communication tool, check to see if
that’s what the child is asking for.
Coping with the challenges of communication
If you feel daunted, frustrated or overwhelmed, try to remember that although
this can be an incredibly dicult task, YOU CAN DO IT. In fact, you have
probably done it already, before your child learned how to speak as a baby.
Although he/she has developed intellectually since that time and now has more
complex ideas to express, remember that with your help, your child was able
to learn a new way of communicating once before and will again. Try to be
patient with yourself, keeping in mind that the diculties you may face with
this new way of communicating are caused by this enormously challenging
situation; always remember that you’re doing the best job you can. If you
need to, take breaks to manage your own stress. Young people can sense your
anxiety, stress, or frustration, so allow yourself the time to refocus and know
that this is a challenging process for any parent.
Communication Strategies
Families have shared with us a range of creative communication strategies they
have used, which fall into two categories:
1. Tools: meaning there is an actual “thing” to help the child express himself.
2. Techniques: referring to a special way of communicating without using a
physical tool.
Please note: this may be an overwhelming list of possibilities. We’ve included
these to assist you in nding what will work best in your situation; you are not
expected to use them all.
Tools
• A bell or buzzer: ese can be used to get someone’s attention if the child is
in a dierent room, or be used as a way to say “yes.”
• Paper and pencil/markers: For kids who have learned to print or write, this
is a familiar way to express their thoughts.
• Magna Doodle: Children can write messages, draw pictures, or draw an
arrow to point the Magna Doodle at what they want. Kids typically enjoy
these because they are familiar and feel like using a toy rather than a “special
device,” and because they are easy to use.
• Laptop/tablet: Children who know how to type like using laptops because
they can send emails or type messages for someone to read while they type.
ey also tend to like that they can watch movies on the same device, although
for some the laptops are too heavy.
• Keyboard: A few children have used regular keyboards that are not connected
to computers. ey press a series of letters to spell a message while someone
else watches and reads what they typed. Special keyboards that have the letters
in alphabetical order can also be used. ese tools help children express a
wide range of messages, but some people who have used the keyboards say
it can take a long time to type messages and a child can forget what letters
they have already typed. Many families create their own keyboards by clearly
writing the
alphabet in large letters on a piece of paper or cardboard for the
child to point to.
• Picture books or boards: ese can be like scrapbooks or a piece of cardboard
with photos or drawings and words, made by family and friends. Children
can point to a picture or word, or parents scroll through, pointing to one
message at a time and waiting for the child to say “yes” when they point at the
right message. Some people nd it frustrating to search for the right message,
especially when the child wants to say something that is not included in the
book or board.
• Feelings faces: A chart showing a variety of faces, including happy, sad, angry,
frustrated, lonely, bored, excited, hopeful, etc., can help children to express
themselves by pointing (or having their parents point) to the feeling they are
having. e number of faces to include depends on a child’s age and abilities;
faces can be added or taken away as a child’s needs change.
• High-tech communication devices: ese devices usually have buttons for
children to press, with each button causing a dierent message to be spoken,
allowing kids to express a range of messages. While some children like using
these, others do not, because certain devices are complicated, seem unfamiliar,
and are sometimes hard to use or learn or feel impersonal.
Tips about tools
• For kids who are able to read, include words as well as pictures or symbols in
Chapter 12: When a Child Can No Longer Speak
202
203
Chapter 12: When a Child Can No Longer Speak
communication tools. Children will associate the words with the symbols so
if it later becomes dicult to read the words, they are still familiar with the
meaning of the symbols.
• Include your child in creating their communication tools (such as books,
boards, high-tech devices) as much as possible. By choosing which images will
represent dierent words, feelings, activities, etc., your child maintains some
control and will feel more connected and involved in the process. Involving
children also promotes familiarity and makes them feel more invested in
using the tools.
• As much as possible, consider your child’s individual voice. For some families
this means recording the child’s voice on a high-tech device or a voice recorder,
saying common messages so he/she can hear his/her own voice. Families who
have done this typically treasure the recording and encourage other families
to do this as early as possible. When such a recording is not possible, families
can record another child’s voice that sounds similar in age and gender. Many
parents have said that it wasn’t just about the sound of the child’s voice but
the kinds of things that he/she would have wanted to say. By including jokes,
sayings or common phrases, a child’s unique personality is able to continue
shining through in a meaningful way.
• If you are using a tool that has preset messages or pictures in it, such as a
picture book or a high tech device, your child may want to express something
that is not included in the tool. In this case, be sure to ask your child about
messages that are not in the tool. Ask, “Is it something that isn’t in here?” to
nd out if that’s the case. en you can use “yes” or “no” questions to nd
out what your child is thinking, and to decide whether or not to add that
new message to the tool.
Techniques
Most families we interviewed said they use special ways of asking questions,
such as oering two choices, asking “yes” or “no” questions, and reading their
children’s body language and facial expressions. Some families also used the
following techniques:
• Signs and signals: ey use their hands or their faces, or adapt sign language,
especially by using the rst letter of a person’s name to refer to that person.
An example of a signal would be a child holding an imaginary cup up to
their mouth to show she is thirsty.
• Pointing: Children can point to things to show what they want, such
as pointing to a window to say they want to go outside. If a child is
uncomfortable, he/she can point to the part of his/her body, or a picture of
a body, to show others where he/she feels discomfort.
• Lip reading: Some children have trouble producing sounds or words but are
still able to make the shape of words with their mouths. For children who
have trouble hearing, a few parents say that by mouthing words slowly, and
exaggerating their mouth’s movements, their children can gure out what
they are saying.
• Physical presence, touch and hugs: When it is too hard to use words,
being close to one another and sharing aection are great ways of expressing
emotions and love.
• Lists: Many parents said it was very important to keep three kinds of lists,
and to keep adding to them, including:
1. Signals, such as what the signals look like and what they mean, (i.e.,
“pointing to mouth means hungry or thirsty”).
2. Common questions that caregivers ask, things that the child frequently
asks or says, or issues or needs that the child has.
3. Clues to a child’s needs, such as body language, time of day, or anything
else that can help caregivers gure out what the child wants or needs.
ese lists may help you remember or think of what to ask, and improve
communication when someone else, who is less familiar with his communication
strategies, is caring for your child.
Tips about communication in general
• When possible, try to adapt familiar communication tools to meet a child’s
changing needs rather than introducing new tools.
• Keep talking to your child. Avoid asking questions that they can’t answer;
stick with “yes” or “no” questions, but keep including your child and asking
her opinions.
• Teach siblings how to communicate using the new tools or techniques. is
helps to encourage interaction and maintain sibling connection.
• Use communication strategies to play games with your child to improve
comfort using the strategies. Children who are able to say “yes” can play
twenty questions; children who are using a communication book can choose
Chapter 12: When a Child Can No Longer Speak
204
205
Chapter 12: When a Child Can No Longer Speak
a message while others try to guess what it is. Play charades by having your
child point out one message for another person to act out.
• Try to be patient with yourself, your child, and the process. ere is no easy
way to do this. Try to stay calm, take deep breaths, and take care of yourself.
Parents and their children can often feel frustrated and helpless. In the midst
of this dicult process, one of the most important things for children to hear
is: “I know that you know what you want to say.”
Challenges
Some children are reluctant to use tools before they are needed and may feel the
tools undermine their current abilities. If a child refuses to use a certain tool, try
telling him/her that he/she does not have to use it right now but that you want
to show him/her how it works anyway. at way if you need to reintroduce it
later, it will be familiar.
While we do not want to force children to use a communication tool they don’t
want to use, if their abilities change unexpectedly and they have not already
had the chance to learn and practice communication strategies, it can be even
more challenging for them to use new techniques to express themselves. For
these reasons, we recommend that families talk about and practice a range of
communication strategies rather than focusing only on one.
Try adopting old strategies as much as possible so your child can keep using
the same approach in a slightly dierent way, rather than learning something
completely new. For example, start with a picture board with many small pictures,
and if your child’s vision starts to change, narrow down the number of pictures,
spread them out, and enlarge them so they’re easier to see. Practicing a variety of
techniques and adapting them (rather than starting something totally dierent)
are ways of helping children feel familiar with dierent communication strategies.
Sometimes you may need to communicate in the midst of a crisis situation or
while your child is distressed; these moments may be brought on by physical and/
or emotional pain that the child is feeling. It is important to know how to calm
yourself and your child so you will be able to work together and communicate
eectively to manage these situations. Practice calming techniques together on
a regular basis. Some examples are deep breathing, blowing bubbles, soothing
touch, or focusing on each other. ese techniques are helpful because when you
and your child are calm, you will be able to communicate more eectively, which
is especially important in an urgent situation.
Deciding what to try
Choosing a strategy for communicating with your child depends upon the child’s
abilities and personal preferences. Consult with your child’s team to nd out what
strategies might be the best suited to your child’s needs, abilities, and preferences.
en, considering your child’s personality, decide which ones to try. Together you
can decide which ones work best. Some children are open to using familiar tools
that feel like play, such as drawing, writing, or using a Magna Doodle. Remember
that the emphasis of communication should be on the connection between you
and your child rather than the content of the messages.
While it is often easier for a child to keep doing what is familiar rather than trying
something new, sometimes there is no choice. If a communication strategy is no
longer working, or if your child is getting too frustrated, it is time for a change!
Communication Topics
Parents we spoke with felt it was important to be able to talk about “everything:”
physical comfort, feelings, worries and “regular conversations” about friends,
jokes, hobbies, and daily activities. Some messages were more concrete—hunger,
discomfort—which are easier for children to express by pointing to a picture or
an object, or answering “yes” or “no” questions. Abstract topics such as emotions,
spirituality, and the future are more dicult to discuss, requiring caregivers to ask
more questions in order to help a child express what he/she is thinking and feeling.
Parents described some of the most important topics they addressed with their
children, and strategies they used to do so. It may seem overwhelming to think
about all of the topics or messages your child may want to express, and the
charts, lists, or strategies you could create. Remember that becoming familiar
with communication strategies will happen over time and with support from
family, friends, and your child’s team at the hospital.
e way you communicate throughout this time will be shaped by your family’s
values, belief systems, personalities, and previous experiences communicating,
especially about dicult topics. While we know sharing information and
discussing feelings helps children and families cope and support one another,
there is no “right” way to go through this experience.
Physical and health needs
Parents described important conversations they’d had with their children about
how their abilities had changed and that the changes would continue. Although
these can be dicult discussions, children cope better when their questions are

Chapter 12: When a Child Can No Longer Speak
206
207
Chapter 12: When a Child Can No Longer Speak
answered than when they are left to wonder and make up their own answers.
To assist with talking with, or responding to your child’s physical needs, below
is a chart [Table: 1] outlining some issues and approaches.
Child’s Messages Parents’ Questions
Ouch/I’m in pain/Something’s
hurting.
“Is something hurting?”/“Are you in
pain?”
If “yes,” ask: “Can you show me where it
hurts?”
Point to dierent parts of your child’s
body, or to a picture of a body, or even
name dierent body parts—“Is it your
head?”/“Is it your stomach?”—and ask
your child to let you know when you’ve
said, or when you have pointed at the
part of the body where he/she feels pain.
Remind your child how to signal “yes.”
I’m uncomfortable. “Are you uncomfortable?”
If “yes,” ask (one at a time, slowly, until
your child indicates “yes”).
“Do you feel sti/numbness/itchy/dizzy/
hot/cold/weak?”
I need to move/I
need to change positions.
“Do you want to move/change posi-
tions?”
If “yes,” ask (one at a time, slowly, until
your child indicates “yes”).
“Do you want to sit up/lean back/lie
down/roll over/move over/sit somewhere
else/lie down somewhere else/go out-
side?”
I need to go to the bathroom. “Do you need to go to the bathroom?”
If “yes,” ask (one at a time, slowly, until
your child indicates “yes”).
“Do you need to use the toilet or a new
diaper/take a shower or bath/brush your
teeth/brush your hair/wash your face?”
I’m hungry. “Are you hungry?”
If “yes,” oer food choices, one at a time,
slowly, until your child indicates “yes.”
Child’s Messages Parents’ Questions
I’m thirsty. “Are you thirsty?”
If “yes,” oer various drink choices, one
at a time, slowly, until your child indi-
cates “yes.”
I need my walker/wheelchair. “Do you want your walker/wheelchair?”
Table 1: Questions and responses
Medical needs
Children we spoke with wanted to know about medical equipment, tests, and
procedures, including the use of dierent medical equipment, how it works, and
what procedures will feel like. When a procedure will be uncomfortable, people
may be afraid of upsetting children by telling them the truth. Unfortunately,
when children are caught o guard by a needle or other unpleasant things, they
do not have the chance to react and then calm down and then try to cope with the
experience before it is time for the procedure. Children may also begin to doubt
caregivers and to think that things are being kept from him even when they’re not.
Children benet from knowing what to expect—where a procedure will take
place, who will be there, what steps are involved, and what it will feel like. is
information gives kids a chance to prepare for what will happen and practice
coping strategies, such as deep breathing, blowing bubbles, holding your hand,
listening to music or a story, using guided imagery, or squeezing a stress-ball.
Children may also benet by watching a simulated procedure on a play-therapy
doll or stued animal like American Childhood Cancer Organization’s Cozy,
the “Port-a-Cat.”
When explaining medical procedures to children, it is important to be honest, to
use language that is clear and simple, and to check in with them by asking “Does
that make sense or would you like me to try to explain it in a dierent way?”
Children may also want to know why treatments are needed, how to know if
they’re working, and what happens if they don’t work. When a child nishes or
stops a certain treatment, he/she may wonder what that means, whether it is
because the disease is gone or because it can’t be cured. ese are dicult concepts
to explain, but if a child has a question, it is better to explore the answers honestly
and openly together than for a child to rely on his own imagination. ese issues
can be overwhelming for children to think about on their own; talking about
them together oers reassurance and support for the child even when there aren’t
clear-cut answers.
Chapter 12: When a Child Can No Longer Speak
208
209
Chapter 12: When a Child Can No Longer Speak
It is helpful to talk to children about what kinds of information they want to be
told about their illness, treatment, and side eects, before communication gets
more dicult. at way you have an idea of what your child wants to know
about and you can continue to provide the information your child wants and
needs throughout his/her journey.
Emotions
To talk about feelings you can use charts with pictures of dierent facial expressions
or use a list of feelings. Your child can point to the face that shows how he/she feels,
or you can point and look for your child to indicate when the right answer is selected.
It is helpful to oer a wide range of feelings so your child can express his/her true
emotions, rather than settling for one that is “close but not quite right.” On the
other hand, if your child is getting overwhelmed, use a shorter list or chart with
four to eight simpler feelings such as happy, sad, scared, mad, bored, etc. Try to
use words that are familiar to your child and make sense given her age. If possible,
try to include your child in creating this list of feelings to ensure she is familiar
with all the words and that she has some control.
Strategies for talking about emotions
Ask your child if he/she feels a certain way. For example, “Are you feeling happy?”
or “You look frustrated, are you feeling that way?” is oers your child the chance
to express an emotion and to oer some control by answering “yes” or “no.”
Share how you are feeling and then ask your child whether he/she is feeling the
same way. is helps a child express his feelings and reassures him/her that others
feel that way, too. However, it is important to recognize that children may feel
dierently than the people around them; this is perfectly normal. Try to say
something like, “I’ve been feeling pretty sad and I wonder if you have, too. You
know, it’s okay to feel sad and it’s okay not to, too.”
Children need to be reassured that all of the feelings they have, no matter how
intense, unfamiliar or conicted, are natural. Let your child know that even though
these are not “easy” feelings to have, they are natural, understandable, and “okay.”
A lot of emotional messages can be conveyed through hugs and touch. Being close
and making eye contact also helps children feel more connected and comforted.
Activities
When a child’s abilities change what he/she is able to do or play with, it is helpful
to have a list of things that your child can do to choose from. Lists also help
parents so they don’t always have to remember all of the options. Some of the
most common activities that parents we interviewed said their children enjoyed
were: listening to music, watching a movie, hearing a story, going outside, playing
a game, writing to someone, making food, or visiting friends.
People and pets
Maintaining relationships with family and friends is very important for young
people. Parents and caregivers can help by giving children a way to ask to see a
special person, or to send them a message. Create a chart with names and photos
of family members, friends, and even pets, for children to point to.
Many children ask about people they know who have died, wondering where they
are now, whether they are “okay” and commenting that they miss these people. It
is natural for a child who has a serious illness to start thinking about life and death
and loved ones who have died. It can be a safe way to wonder about these things,
an indirect way for children to show you that they’re thinking about death, and a
way to start a dicult conversation. Also, when children realize their loved ones
are still remembered and loved after they’ve died, it oers them the reassurance
that they, too, will be remembered and loved after their death.
e Future and Spirituality
It is natural for children to wonder about these topics, especially as they feel their
bodies changing and sense the emotions in the people around them. It can be very
hard for children to initiate conversations, especially when they fear that talking
about these things will be upsetting for others. ey may ask questions in indirect
ways, such as asking about the death of a pet, or the death of someone else, or
general questions about what happens after you die. Because of how dicult it
can be for children to bring up these topics, it is very important to support them
when they want to have these conversations, rather than avoiding or changing
the subject.
If your child has questions about death and spirituality, try to answer his/her
questions as honestly, clearly and calmly as you can. He/she may ask you questions
you don’t have answers to. at’s okay. You can say you’re not sure, that many
people wonder about questions like that, and that it’s okay to wonder about these
things together, even without nding any answers.
In interviewing parents, some of the biggest struggles they said they faced were
about whether or not to tell a child that he/she could or would die and how to do
that. Research and our own clinical experiences suggest that children and families
benet from having open and honest conversations. Families who do this said
Chapter 12: When a Child Can No Longer Speak
210
211
Chapter 12: When a Child Can No Longer Speak
they did not later regret having had these conversations. Many parents said that
even though it was so hard for them to talk about these things, after they had
spoken with their child about death or spirituality, they realized the child seemed
comforted and relieved, and that they—as parents—did as well. Whether you
decide to talk with your child about death and spirituality and how you do this
is up to you and will be a very personal decision based on your experiences and
your beliefs.
About the future
For some children, thinking about the future can include writing a will, exploring
organ donation, or planning a memorial celebration. Parents often worry that
talking about these things with their children will cause them to lose hope. On the
contrary, if a child is already thinking about these things, the opportunity to share
his/her thoughts and feelings about them with loved ones can oer tremendous
comfort and relief, a sense of control, and the opportunity to plan his/her own legacy.
You can also talk about how you will remember and honor your child at holidays,
family events, birthdays, and other special times. Some families have a special meal
or celebration, wear a special piece of clothing or jewelry, listen to a certain song
or musician, or make up their own unique rituals for these special times. Others
may plant a memorial tree or garden, hold a fundraiser, or create a scholarship
in the child’s name. Some children have their own ideas about how they want
their family to remember them, and many children want to be involved in family
discussions about this. Not only does it reassure the child that he/she will not be
forgotten but it gives him/her a clear idea of exactly how her family will remember
and feel connected to him/her.
About spirituality
Some children ask their parents questions about what happens after someone
dies, what they will do when they are in heaven, or how their families will feel
their presence. Whatever your beliefs are, you can share them with your child.
Many people don’t know what they believe, or may believe that there is nothing
after death. If this is the case for you, you can explain to your child that many
people have dierent beliefs and that you’re not sure what will happen, or that
you’re not sure whether any of them will happen; either way, ask your child what
he/she thinks, or would like to think.
Regardless of what you believe about what happens after death, you can talk with
your child about how he/she will always be part of your family even though he/she
will not be physically present. ings he/she taught others, personality traits, his/
her values, and hobbies that he/she shared with others, are all deeply meaningful
ways that his/her life will continue to impact his/her loved ones.
Caring for Your Children and Yourselves
is is a very dicult and challenging experience for parents and children.
Developing strategies to manage the impact of the ongoing loss of abilities
can be as important as developing communication strategies. is section is
meant to assist parents and caregivers in thinking about and addressing some
of these challenges.
Supporting the child with a brain tumor
Here are examples of some of the challenges and concerns that parents described
and the things that can help kids deal with them. Strategies from the previous
sections will assist in dealing with these issues.
Feeling frustrated
When a child nds that he/she can no longer do something that used to be
easy to do, or realizes that so much about his/her body or his/her life is beyond
his/her control, frustration is a natural reaction. e loss of independence or
needing help with things such as eating or going to the bathroom can be very
upsetting, especially as children realize they will not regain the ability to do those
things on their own. is kind of frustration might be expressed in dierent
ways, such as being impatient or getting angry. One way to help children cope
with these feelings is to help them nd ways to express themselves with words
added to a communication board or book, or physically using a stress ball made
out of Play-Doh
.
People often want to cheer kids up when they are feeling upset; sometimes they
try to distract them by talking about something fun or focusing on an activity.
But when children have these strong feelings, they need ways for their feelings
to be expressed and heard—and to know that someone else understands—before
they are ready to move beyond these emotions. It’s important to be patient and
let your child know that you will work together to gure out what he/she wants
or needs, whatever it may be.
Feeling self-conscious
As their bodies and their abilities change, it is common for children to feel less
comfortable around others. Children, particularly teenagers, are often fearful
about being seen as “dierent” or being treated “dierently” than others.
Chapter 12: When a Child Can No Longer Speak
212
213
Chapter 12: When a Child Can No Longer Speak
Educating a child’s peers about his/her illness, explaining that a tumor is not
contagious and that it is the reason for his/her changing abilities, and helping
them learn useful communication techniques can be a very good way to help
them understand and relate to one another. ere may be someone at your
child’s hospital—such as a nurse, a child life specialist, or a social worker—
who can visit your child’s classroom to talk about these things. Teachers and
other school sta are often very helpful in organizing this kind of classroom
experience. On the other hand, some children feel strongly that they do not
want other people to know about their illness and would not be comfortable
having someone speak with their classmates. Sometimes it helps to talk with
your child about what he/she is afraid would happen if others found out, and
you may be able to dispel these fears and facilitate the connection.
However, if your child does not change his/her mind, it is important to respect
his/her wishes in order to avoid your child feeling embarrassed, helpless, or even
vulnerable. ere are so few things that a child in this situation can control that
deciding what information to share with others may be one of the few things
that he/she can control.
A few parents described their children feeling self-conscious about
communication. MSN and other online chat systems, email, social networking
sites, text messages, or even written letters can be great ways to help children
keep in touch with their friends without having to feel so self-conscious. Also,
if your child is comfortable with you teaching others how to use the specic
communication strategies your family has developed, with time and practice,
his/her feelings of self-consciousness may decrease.
Missing familiar people and activities
Familiarity provides so much comfort to children. When it’s possible to help children
continue to participate in these kinds of activities, even if it means participating
in a dierent way than they used to, it can be very helpful for them. On the other
hand, some children may nd that there are some things they don’t want to continue
being involved in. If this is the case for your child, try to help him explain why
he feels this way. It may be that he/she is self-conscious and afraid of how others
might treat him/her, in which case you can talk to him/her about anything that can
be done to help make the situation more comfortable or inviting. In some cases a
child may feel uncomfortable or even unsafe in dierent environments. Whatever
the situation, respecting your child’s wishes as much as possible will help him/her
feel more comfortable and safe and give him/her a sense of control.
Coping with medical experiences
Play is a great way to help children cope with dicult experiences. In times
of stress, play may be the furthest thing from our minds, but it may also be
the most valuable tool. Blowing bubbles, bringing paper and crayons to draw
or play tic-tac-toe, a deck of cards, or even a list of games such as “I Spy” or
“Twenty Questions” are all simple and useful distractions. For older children
and teenagers, think back to what has helped them before; listening to music,
playing a video game, or reading a book may be useful distraction techniques.
Guided imagery, deep breathing, and other relaxation techniques can also help
children of all ages cope with anxiety related to medical issues. Talking with
your child about what is happening, what medical procedures might feel like,
and any other questions or concerns they might have will help them better
manage these experiences.
Knowing they will be cared for
Parents highlighted that it was extremely important for their children to know
that they would be well cared for. is concept included three things.
1. Knowing that the health care team would continue to care for them. When
they know that a disease or a tumor is not curable, children may think that
means there will be no more medical care.
2. Knowing that they will still be looked after and that their pain and other
symptoms will still be managed is very important.
3. Knowing that they are not alone and that their parents and their family will
always be with them and love them “no matter what.” When children are
struggling with how they’re feeling and the ways their bodies are changing,
this may be the most valuable comfort you can oer them.
Children’s concern for others
Another common and important concern parents told us about is children’s
worries about whether their parents and their families will be okay after the child
dies. Parents said it is very important to address these concerns by letting your
child know two things: that the family will be sad and will miss the child after
he/she dies, but at the same time, the family will be alright. Families did their
best to try to ease the child’s burden of worrying about how his/her loved ones
will cope. It’s important to express one’s love for the child while acknowledging
the impact of his/her loss.
Chapter 12: When a Child Can No Longer Speak
214
215
Chapter 12: When a Child Can No Longer Speak
e importance of communicating
Parents told us that they often feel helpless and frustrated that they are not able
to change the situation and protect their child from what is happening. Of course
this feeling is natural. Sometimes in an attempt to protect a child, parents avoid
talking with their child about his/her illness or letting him/her know that he/she
is going to die. Although this is done with the best intentions, it does not have
the impact parents hope for. Some of the unintended, possible consequences are:
• When children are not invited to talk about their illness, they learn from
others’ example not to raise the issue themselves. Without having someone
to talk to about their thoughts and worries, they are left to wonder on their
own, using their imaginations to answer their own questions.
• Children are very sensitive to the emotions of the people around them and
know when others are upset. ey can recognize when something is being
kept from them and can only wonder what that might be, often imagining
the worst.
• Children are more aware than anyone of the changes occurring in their own
bodies. Although they may not know what will happen in the future, they
have learned that unpredictable changes can continue to occur. If they do
not feel able to talk about their illness or the future, they are left to face
these questions and fears on their own.
With these things in mind, it is clear that protecting a child from talking
about his/her illness does not protect him/her from the dicult experience he/
she is already living. Instead of letting this fact make you feel helpless, try to
see that it actually oers you an important opportunity. You are not helpless.
Even though no one can change what is happening, there is a great deal that
you can do to help your child through this experience. As we’ve discussed in
this section, there are some very important messages that will oer your child
comfort, reassurance, and security. Make sure your child knows the following.
• Your child is not alone. You will be there to support him/her throughout
this experience.
• Your child can trust you. You can truthfully prepare him/her for things
such as medical procedures and other events so he/she feels less anxious
and surprised by these things. Your child’s health care team at the hospital
can help you gure out how to do this.
• Your child will be well cared for. You can reassure your child that you, your
family, and your child’s health care team will all be working to make sure
that he/she has what he/she needs to feel comfortable and taken care of.
• Your child will always be part of your family. You can talk about all of the
things you will remember and all of the ways that your child will continue
to have an important place in your family.
• Your family and people who know and love your child will be incredibly
sad when he/she dies, but your family members and friends will support
one another through their grief.
Although these things cannot change what is happening to your child, they can
make him/her feel supported in the knowledge that he/she will not be alone.
Nothing can take away the pain that your child and your family will struggle
with, but these important messages can oer your child support and strength
as you face what is happening, together.
Supporting siblings
As a parent, you may not only be supporting a child who has a DIPG but also
his or her siblings. ere are some issues that are common for children who
have a sibling living with a serious illness, and these can vary depending on the
age of the children. For example, many children in this situation have questions
about why this happened, worries about their own and/or their sibling’s health,
and concerns about their parents’ emotional struggles. It is also very common
for children to wonder if they are somehow to blame for a sibling’s illness and
to worry that they may also “catch” the illness. Even if a child has not expressed
these worries, it is helpful to say something like, “I just want to make sure you
know that there is nothing you could have done to make this happen and that
this is not the kind of illness you can catch from someone else.”
Siblings may also have questions about the future. e suggestions in this chapter
about how to talk with a child about his or her own illness and the future, as
discussed in the previous sections, also apply to talking with the child’s siblings.
Many of the parents we spoke with shared their suggestions about how to help
brothers and sisters.
• Make sure the siblings are able to continue spending time together at home
or in the hospital.
• Help all of your children learn how to use the new communication
strategies, as it can help children continue to interact with each other and
maintain their relationships.
Chapter 12: When a Child Can No Longer Speak
216
217
Chapter 12: When a Child Can No Longer Speak
• Encourage siblings to say “hello” and “goodbye” to their sibling when they
come home and when they go out.
• Encourage interactions that help a sick child continue to feel recognized and
included in the family’s day-to-day activities despite their changing abilities.
Brothers and sisters may be reluctant or nervous about learning new
communication strategies, and may be afraid of doing it “wrong” or looking
silly if they do. Just like teaching communication strategies to a child who is
sick, it can also help to use games to teach these strategies to their siblings,
and to practice with them until they feel more comfortable using them. ey
may also need your help to understand why their brother or sister isn’t able
to talk the way they used to. Because they may not be able to see any physical
evidence of something stopping their sibling from being able to speak, some
children wonder why their brother or sister just doesn’t try harder. It helps to
explain that our brains are like computers that send signals or instructions to
all of the other parts of our body to make it work, including our arms, legs,
stomach, heart, lungs, eyes, ears, mouth, etc. When a person has a brain tumor,
it interferes with, or “mixes up,” some signals so that things don’t always work
the way they’re supposed to. is is why some children who have brain tumors
aren’t able to speak the way they used to.
Similarly, children may not know how to interact or play with their brother or
sister since their abilities have changed. ey may also believe that their sibling
doesn’t want to play with them anymore. Again, it is important to explain that
these changes are caused by the tumor rather than being the child’s choice.
en you can help your children nd new ways of playing or being together.
Healthy siblings can read stories to their brother or sister, watch movies or listen
to music together. ey can also play “for” their sibling; some examples of this
are making a beaded bracelet or building a LEGO tower by asking their sibling
what color bead or LEGO block to use next. ey can also draw a picture or
write a story based on their sibling’s ideas about what to draw or write. When
thinking about how to help children play together, consider what they used to
do together and try to nd ways to adapt those activities. Children may have
a hard time trying new things; it can be easier and more comfortable to do
what feels familiar.
Some other considerations we’ve learned about siblings are:
• Healthy siblings need opportunities to play for themselves.
• ey will need your assistance to nd a balance between feeling helpful
without taking on too much responsibility for their sibling with a brain
tumor.
• Even when they understand why their brother or sister needs the extra
attention, siblings need support to make sense of, and express, their
emotions and possible feelings of jealousy about the extra attention their
ill sibling is getting.
• Sometimes siblings are asked to be patient, helpful, and understanding for
a long time, which isn’t easy. is is a challenging experience for children
of all ages, and their frustrations can be expressed dierently at dierent
developmental stages.
• All children need to know that their needs will be met.
• Children of all ages need love and support from their parents, though how
they express this need changes at dierent ages.
• It is important to recognize and tell each child how much you appreciate all
that he or she has done throughout their sibling’s illness, including specic
examples when possible.
• Let them know that you recognize how challenging it has been and will
continue to be and encourage them to let you know when they’re struggling
and need help.
Talking with your other children about how they are feeling, helping them to
understand that all of their emotions are natural, and encouraging them to
express any questions or fears that they have is very important and benecial.
ere may be people at the hospital or at school, such as child life specialists,
social workers, counselors, or volunteers who can help support children when
their sibling is ill. ere may also be local organizations that can provide support.
Parents’ Advice for Other Parents
e parents we spoke with shared some very personal insights into their
experiences that may be helpful advice for others parents. Some of these are
reections or quotes about a parent’s outlook or important things that they
tried to keep in mind while going through this same process with their child.
About relating to children
• Know your child, their personality, interests, coping styles, and preferences
for support.
Chapter 12: When a Child Can No Longer Speak
218
219
Chapter 12: When a Child Can No Longer Speak
• Have important conversations sooner rather than later. Have important
conversations about topics such as illness, life, death, your love for them,
and spirituality as early as possible. Although these can be emotionally
dicult conversations to have, they get even more dicult once children
have a harder time communicating.
• Keep communicating. When a child can no longer express themselves to
others, it can be hard to know whether or not to continue to talk to them.
Communicating through story-telling and touch (including a hug, gently
squeezing or rubbing a child’s arm) can convey love, warmth, aection,
and provide great comfort to a child.
About relating to one another as parents and as a family
Try to work together—as a couple, as parents, and as a family.
Asking questions and asking for help
• Whatever you want to know, ask. If there is anything you have wondered
or worried about, do not hesitate to ask a member of your child’s health
care team.
• Whatever you need, ask. Dierent services will be available depending on
the hospital or the community where you live. Ask a member of your child’s
health care team to help you nd resources near you.
Acknowledgements
We want to express our sincerest thank you to the families who participated
in this research and who shared their experiences and insights. ank you to
our colleagues, Dr. Eric Bouet, Dr. Ute Bartels, Cindy Van Halderen and
Dr. Tom Chau. Funding for this research and the creation of a handbook for
families and caregivers has been generously provided by B.r.a.i.n.child at www.
sickkids.ca/brainchild.
Parent Perspectives
Caleb was diagnosed with DIPG January 28th, 2010. At the time of
diagnosis his speech was slightly slurred. Within a week of starting steroids
and radiation this cleared completely until progression. In December 2010
we noticed his speech became very nasal; progression was confirmed
January 13, 2011. Steroids and a second round of radiation helped but
not completely.
In June 2011 he began having a lot of trouble with speech, to the point
that no one, other than his father, his sister and me could understand 75%
of what he was saying. Caleb was nine and a half years old at the time.
This frustrated and upset him far more than any other symptoms, including
the loss of strength in his left side and the ability to walk. He told us he
could hear himself perfectly clear and could not understand why no one
could understand him. He said that if he could hear what we heard then he
would know how to try to fix his words, but since he heard himself "clear
as a bell," he had no idea how to help us understand. This was extremely
frustrating for him.
Initially he would speak one word at a time and we would repeat what we
thought he had said until we guessed what that word was before moving
on to the next word. Caleb was very witty—a funny little man who had the
best “one-liners,” so it saddened him so much when he would try to speak
and no one understood. He told me that things are not funny when they
have to be repeated over and over. That was when he began to withdraw.
We bought a small white board that we carried everywhere we went so
he could write down whatever he wanted to say. It helped, but he missed
talking and being part of conversations. When he spent time with friends
he would become so sad because whenever he wanted to say something he
would begin to write but by the time he was done writing, his friends had
moved on to a new topic. That is when we insisted on more one-on-one visits
instead of group visits with friends and family. This helped a lot because
he didn't feel he had to compete to get his thoughts across and he didn't
lose the chance to have his point heard.
As time went on and he began losing the ability to write (some days he could,
Chapter 12: When a Child Can No Longer Speak
220
221
Chapter 12: When a Child Can No Longer Speak
and some days he couldn't) he would lose his interest in communicating and
he didn't want those around him talking or communicating either. We found
it very important to match his mood. If he was in a good mood it was okay
to laugh and talk a little, but when he wasn't we tried to keep the house
quiet and low key. Up until this point his relationship with his sister, Avery
(seven and a half years old at the time) had held strong, but once Caleb's
ability to communicate had diminished he did not want her around much
and she avoided him as well. She learned very quickly to only ask “yes or
no” questions and she would always ask if she could tell him something
instead of bombarding him with her conversation. Giving him that choice
helped him feel some sense of control. This helped with his patience and
his willingness to listen to her.
We also made up small cards with words on them so he could pick out one
or two that would help us guess what he was thinking. In the end when he
was not able to move at all we wrote the letters of the alphabet on a white
board, we would point to the letters and he would either nod or blink when
we had the right letter, then we would move on to the next letter, spelling
each word out. It was extremely slow but we only used this method when
he needed something new or out of the ordinary.
Throughout the process of Caleb losing his ability to communicate, I become
acutely aware of his needs. I learned his expressions and could usually
guess fairly accurately what he wanted or needed through them. All of this
was extremely hard for all of us to adjust to, but with these tools we were
able to keep as much fun in his days as we could.
When Ella started to lose her ability to speak, we created a picture book
with photos of all sorts of things she liked to do, needed to do, and feelings.
When we couldn't understand her speech we would pull it out and flip the
pages. She had just enough strength to point to the pictures. My favorite
was the “I Love You” picture because I missed hearing her sweet voice
say those words to me.
Warren’s speech wasn’t very good. People had a very hard time
understanding him. I had a hard time too. He would have to repeat things
a lot and he would get frustrated. Other than listening hard I didn’t know
what to do. Looking back both of us learning some simple sign language
such as drink, eat, bathroom might have helped.
Whereas in August and prior, Stella was speaking in full sentences with a
huge vocabulary, now it takes her up to 30 seconds to squeak out one word,
which is generally difficult if not impossible to comprehend.
We find it necessary to be close to him and looking directly at him to
understand his speech. He is not at all frustrated by this and patiently
repeats himself as often as necessary so that we know exactly what he
wants to tell us.
It was Monday, Nov. 29th, when our 4-year-old Julian woke me at 5 a.m. I
noticed he wasn’t nishing his sentences as he kept repeating, “Mommy, I just
wanna... I just... Mommy, I just wanna...” Crediting the early hour and perhaps
a still sleepy state, we pulled him into our bed to sleep a little longer. When he
woke again, this time vomiting, lethargic and still speaking as if confused, we
knew something was terribly wrong.
Three months after that dreaded day and diagnosis, while searching for signs
of hope on the internet, I came across a site lled with text by a father who
had lost his daughter. He detailed DIPG treatment options and what to expect
during “end of life” care. I read a passage about how most children lose the
ability to speak in the last month or so due to tumor progression or, in the
author’s daughter’s case, a “stroke-like” episode. I recall wanting to throw my
computer across the room. Julian was famous for his sweet, raspy voice and
endless chattiness. Our conversations were treasures and I could not bear the
thought of not hearing his voice let alone him not being able to tell us what he
needed or wanted.
A few weeks later, Julian did suffer a series of seizures that left his speech
slowed but intact. In his nal two weeks, he spoke less and less, but was still
able to point and nod in answer to our questions. We were fortunate in that he
still shared a few beautiful words right up until his last day.
Like many children with DIPG, Caleb was unable to speak during the final
days of his life. At first, we developed a system of communication involving
Chapter 12: When a Child Can No Longer Speak
222
both side to side and up and down eye movement. Then, he lost the ability
to even move his eyeballs from side to side. After that, up was “yes” and
down was “no.”
We were at home, with Caleb set up in a hospital bed in the living room. His
school principal (who had been his 5th grade teacher) contacted classmates
she knew would want to see him and established a visitation schedule. The
children were precious. They sat beside his bed and talked to him, read to
him, remembered. The memory of his best friend sitting beside him on the
bed is seared into my mind. His buddy was scared, unsure what to say, so
I was trying to help. I explained something I’d been doing and concluded
with, “but that gets on Caleb’s nerves.” Immediately, Caleb’s eyes began
moving up and down, up and down forcefully. He was clearly saying “YES
IT DOES!” What a gift that even in those final hours and with such limited
abilities, he continued to bless us with his quick wit and sense of humor.
He helped his best buddy realize that even though his body was no longer
cooperating, our Caleb was with us at that moment.

223
Chapter 13: Overcoming Research Hurdles in DIPG
Chapter 13
Overcoming Research
Hurdles in DIPG
Patricia Baxter, MD
Susan Blaney, MD
e treatment of childhood brain tumors remains a tremendous challenge for
pediatric oncologists, particularly the treatment of aggressive brain tumors such
as high-grade gliomas (e.g., anaplastic astrocytoma or glioblastoma multiforme
[GBM]). is challenge is even greater when the tumor is located in an area of
the brain that is not amenable to surgical resection, such as the hypothalamus
or brainstem (pons). e subsets of pediatric glial tumors that are located in the
brainstem are also known as brainstem gliomas (BSG), or diuse intrinsic pontine
gliomas (DIPGs). As discussed in previous chapters, the diagnosis of DIPG is most
commonly made by a radiologist after reading the magnetic resonance imaging
(MRI) scan, rather than by a pathologist after a neurosurgical procedure such as
biopsy or tumor resection.
So how do we know that DIPGs are high-grade glial tumors if there is no biopsy
or other surgical procedure to determine the tumor pathology? And because
treatment progress for many other types of cancer has been the result of laboratory
studies of human tumor tissue, how can scientists advance research on this type
of tumor and develop eective treatments for children who have a DIPG without
tissue from a biopsy or surgery? ese important questions will be addressed in
this chapter.
DIPG Research Hurdles
Physicians and scientists are optimistic that with modern scientic tools, progress
will be realized in the treatment of children
with DIPGs. Patients, parents, physicians, and
scientists must all work together to overcome
the research hurdles associated with DIPGs,
Dr. Baxter is an Assistant Professor
of Pediatrics at Texas Children’s
Cancer Center, and Baylor
College of Medicine, Houston, TX.

Chapter 13: Overcoming Research Hurdles in DIPG
224
225
Chapter 13: Overcoming Research Hurdles in DIPG
one of the most, if not the most challenging childhood tumor.
Biopsy tissue challenges
e diagnosis of a DIPG is particularly dicult for many parents to accept because
tumor biopsies with resulting pathology are not routinely performed for DIPGs in the
majority of pediatric oncology centers. Surgical resection of DIPGs is not performed
because the brainstem serves as the main roadway for all information that travels to
and from the brain. e brainstem is critical for vital functions such as breathing,
maintaining blood pressure and heart rate, along with other physiological functions
essential to life (see chapter 3). DIPGs are physically located within the brainstem,
and the tumor itself is intertwined with the non-cancerous cells that conduct these
vital life functions. ere is not a denitive border that distinguishes the tumor from
the normal brain tissue, so DIPGs cannot be surgically removed.
Several decades ago, neurosurgeons routinely performed diagnostic biopsies on
children with symptoms of a DIPG and found that these tumors were, with rare
exception, high-grade glial tumors. With the advent of improved imaging technology
and the widespread availability of MRI, it was found that a diagnosis of DIPG could
reliably be made through a history, physical examination, and MRI. As a result, the
practice of diagnostic biopsy was abandoned because of the small, but very real,
risk of severe or life-threatening complications associated with a biopsy and the fact
that past biopsy results have not changed treatment recommendations. is lack of
available tissue from biopsy specimens however, creates an enormous research hurdle
for investigators who need tumor tissue to perform essential molecular analyses of
this unique tumor.
In the future, physicians may routinely recommend biopsy for children with a
clinical and radiographic diagnosis of DIPG. Currently, this recommendation only
occurs when a child’s physician believes the potential benet of a biopsy outweighs
the potential risks, for example, if the MRI ndings are not classical for a DIPG.
In the future, the most likely situation in which biopsies would be recommended
would be if scientists were able to analyze the tumor tissue and identify a set of
tumor genes or other characteristics specic to the tumor that could suggest one
treatment may be of greater benet to a child with a DIPG than another. In this
case, a physician and parent would want the
child to receive the treatment that has a higher
likelihood of benet. Scientists make constant
advances through research that have made
such scenarios a reality for a variety of other
tumors. Our hope is that such advances will
occur sooner rather than later for DIPGs. However, this requires ongoing intensive
research by the best scientic minds and access to tumor tissue, which will be
discussed below.
Challenges associated with the tumor location
Progress has been made in the treatment of some high-grade glial tumors, particularly
those that can be surgically removed. However, progress has been slower for tumors
that cannot be surgically removed, such as DIPGs. As noted above, the location
of DIPGs deep in the brain impedes ready access by the surgeon to tumor tissue.
Tumor tissue is essential to the work of research scientists who are trying to acquire
a better understanding of the tumor’s basic biology and unlock the key for guring
out how a DIPG rst develops. is understanding is needed for the development
of better treatments for this disease. Once scientists understand the tumor’s biology,
they can develop strategies to destroy the tumor cells or to convert them to cells that
no longer behave in a malignant fashion.
Tumor cells can be destroyed in a variety of ways. However, the location of a DIPG
within the central nervous system (CNS) makes all potential treatment approaches
even more challenging, because the CNS has natural mechanisms (the Blood-
Brain-Barrier) that isolate it from other parts of the body. is isolation protects the
brain from damage or side eects associated with chemicals in the blood, including
chemotherapy. Yet this isolation also means that most anti-cancer drugs in the blood
do not get into the central nervous system to any appreciable extent. In everyday
life this isolation is good, because it protects the brain. But this isolation is a major
obstacle in the treatment of brain tumors because the target of the therapy is in the
CNS. us, researchers are developing strategies to ensure drug delivery to the tumor.
is can be accomplished by delivering a drug directly into the CNS, by making
drugs that can more readily enter the CNS, or by developing special carriers (e.g.,
nanoparticles) that can deliver the anti-cancer drug to the tumor.
Clinical trial challenges
Despite the many challenges in treating DIPGs, doctors and scientists, assisted by
patients and their families, continue to work diligently toward nding new and
more eective therapies for children with DIPGs and other brain tumors. While
progress has been slower than desired, we continue to make incremental advances
in understanding the biology of these challenging cancers. While we are working to
unlock the keys to determine what makes a DIPG develop, we are simultaneously
evaluating new drugs in clinical trials to determine whether or not they should be
used in the treatment of DIPGs.
Dr. Blaney is a Professor and
Vice Chair for Research in the
Department of Pediatrics at Texas
Children’s Cancer Center, and
Deputy Director, Baylor College
of Medicine, Houston, TX.
Chapter 13: Overcoming Research Hurdles in DIPG
226
227
Chapter 13: Overcoming Research Hurdles in DIPG
Because the survival rate for children with DIPGs is unacceptable, doctors and scientists
are constantly evaluating new drugs, biological agents, and immunotherapeutic
strategies to improve survival. e best way to evaluate new treatments is through
clinical trials. Clinical trials are a scientically rigorous way to determine the best dose
and schedules of new agents and treatment strategies, and to ultimately determine
whether a new treatment is of benet for a particular disease. As such, many patients
with aggressive cancers such as DIPGs are oered the option of a clinical trial either
at diagnosis or relapse, if a trial is available.
Local radiation to the brainstem is the standard treatment for children diagnosed
with a DIPG. Because the benets of radiation in DIPGs are temporary (typically
less than 1 year), and because there are no chemotherapy agents that have a known
benecial eect for DIPGs, many of the current clinical trials for children with a
DIPG are early phase clinical trials (Phase 1 or Phase 2) where the goals of therapy
are to: a/ nd the appropriate dose of medication to give with radiation therapy;
b/learn more about the side eects of the medications when given with radiation
therapy; and c/ nd early information about the eectiveness of a new treatment.
ere are many contributing factors associated with clinical trial participation
that create additional hurdles for DIPG research. Not all patients will qualify for
participation in clinical trials. All clinical trials are conducted using strict guidelines to
minimize the risk to patient safety. Patients must also have a physical and neurological
examination, laboratory tests, and imaging studies (scans) to determine if they are
eligible to participate in the trial. ese additional tests and the clinical trial itself
might be regarded by the family as interfering with the quality of life of their child.
Trial requirements may also result in disappointment or frustration if a child is not
eligible to participate or if no open slots are available for trial enrollment.
e decision to participate in a clinical trial, if eligible, is also a personal one. e
decision may be inuenced by the potential for benet from the treatment; the
potential side eects of the treatment; a need to travel to a new treatment facility that
is far from home; a desire to help children in the future; and other factors. When
balancing the pros and cons of participation, the family must decide what is best for
their child. is might mean that participation in a clinical trial is not the decision
that they choose to make.
Clinical trial phases and institution challenges
Clinical trials are normally divided into three phases––Phase 1, Phase 2, and Phase
3. Phase 1 trials are most frequently performed in a limited number of pediatric
oncology centers that have specially trained clinical research personnel. In North
America, the institutions that perform these studies are comprised of a small group
of institutions that have been designated by the National Cancer Institute to work
together to perform these trials. ese groups include the Children’s Oncology Group
Phase 1 and Pilot Consortium (comprising 20 pediatric treatment centers) and the
Pediatric Brain Tumor Consortium (comprising 11 pediatric treatment centers). e
formation of these consortia facilitates patient access throughout the country to a
treatment center closer to home and allows trials to be conducted more eciently.
After the optimal dose and schedule of the new therapy have been determined in
Phase 1, Phase 2 trials are conducted to evaluate whether or not the new treatment
is eective for children with DIPGs. Phase 2 trials are typically conducted by the
Children’s Oncology Group, which is comprised of more than 200 children’s oncology
treatment centers in North America. At this stage, access is more widely available
to children throughout the United States. Because the Phase I trials are conducted
at fewer sites, this might make participation dicult for many families. Traveling
distances to participate in a trial can seem overwhelming when there is no clear benet
from a Phase I trial for the participating child. Additional mobility challenges, as
well as communication issues impacting consent and assent that are common with
DIPG children make participation in clinical trials even more dicult. Delays to
clinical trial enrollment prolong the time to completion of the study and analysis of
the trial to determine the ecacy of the new therapy being tested.
Benets, risks, and accrual challenges of clinical trials
ere are benets and risks of participating in a clinical trial. is too can impact
a parent’s decision to not enroll their child into a study, leading to delays in study
accrual and research progress. Participation gives patients access to new therapies
that are promising, but the best dose is often not yet known and side eects may be
present without ultimate direct benet for the patient. However, all clinical trials lead
to information that helps future patients. e dramatic success in the treatment and
cure of other childhood cancers such as leukemia, lymphoma, Wilms tumor, and
many other types would not have occurred without the participation of patients and
physicians in clinical trials. All clinical trials in children with cancer have scientic
rationale and the potential for benet, but no guarantee of benet. is is a challenge
to families as they are faced with the decision of participating or not.
Another challenge to progress in the treatment of DIPGs is the length of time it takes
to complete a trial. DIPG is a rare tumor and it may take several years to accrue an
adequate number of patients for a trial. A drug or therapy may sound very promising
based on information about its use in other tumor types or in an individual patient,
but to determine whether or not there is an actual benet for patients with a DIPG,
there needs to be a scientic evaluation through a clinical trial. If children are treated
with new therapies at random—meaning outside of clinical trials—there is risk of
Chapter 13: Overcoming Research Hurdles in DIPG
228
229
Chapter 13: Overcoming Research Hurdles in DIPG
exposing them to serious side eects, without the benet of learning whether the
treatment is eective and safe. To be scientically valid research, there needs to be a
sucient number of patients entered into the study to determine the benet of any
therapy. Given such a small patient population, it takes time to accrue the required
number of patients in order to give denitive scientic validity to the study. is too
can be a dicult research hurdle to overcome.
In the future, we hope to have enough understanding of DIPG biology to tailor the
treatment for each individual child based on an analysis of a tissue sample (biopsy)
from his or her tumor. We do not have that knowledge today, and such knowledge
will only come from rigorous scientic studies, which includes clinical trials.
How Can the Barriers be Removed?
As already noted, many barriers exist to progress in the research and treatment
of DIPGs. Because of the tumor’s location, surgery and biopsy are generally not
performed. e resultant lack of tumor tissue from patients at diagnosis limits
researchers’ ability to understand the biology of these tumors. Scientists must
therefore make inferences from what we know about similar tumors in adults
and children in our treatment approaches, which may or may not be appropriate.
So, how can progress be realized?
Parents of pediatric cancer patients are the advocacy voice of their children. It has
been through the initiatives of parent advocates that progress has been realized in
many types of childhood cancer treatment. In the early 1970s, a group of parents
of children with cancer formed Candlelighters Childhood Cancer Foundation
(now the American Childhood Cancer Organization) and lobbied Congress for
childhood cancer research funding. eir eorts led to 1) an increase in awareness
of the devastation of childhood cancer, 2) designated pediatric oncology program
funding within the National Cancer Institute (NCI), 3) the inclusion of pediatric
oncology language in President Nixon's National Cancer Act of 1971, and 4)
the development of information and support programs within Candlelighters.
Similarly, parents of children diagnosed with DIPG have a tremendous
opportunity to make a dierence in the future outcome of this disease. A number
of families have responded by forming non-prot organizations in memory
of their children. Some of these organizations provide funding for promising
research, some oer nancial help to families, some have websites populated
with invaluable DIPG specic information, and others raise awareness of this
disease in the community and amongst national funding agencies such as the
federal government and the NCI. Individual parents have dedicated their time to
the creation and administration of online groups in an eort to provide support
and allow families to connect with one another. All of these eorts contribute to
progress toward making life better for children with DIPG.
For some parents, donation of their child’s post-mortem tumor tissue is a way to
make a dierence. is seless act provides tissue that would not otherwise be
available for research. As a result, scientists have recently been able to establish
DIPG cell lines and animal models, and to advance their understanding of the
biology of these challenging tumors. It is hoped that this progress will make it
possible to quickly develop, test and prioritize DIPG treatments for evaluation
in clinical trials. Tissue donation is perceived by many families as a way for their
child to nally be free from the ravages of DIPG, while giving hope to those
children who will follow in their path. Sandy Smith and Kimberlee Spady are
two DIPG parents who, along with others, have devoted themselves to providing
families with information regarding, and/or assistance with, the planning of
autopsy tissue donation. It is their goal to ensure that once a family has made
the decision to donate tumor tissue, they will not be burdened with the details
of arranging the donation. ey also oer support to families from the time of
diagnosis, helping with treatment information, and hoping for the best possible
outcome for each child.
Individuals and foundations are working with determination to raise much needed
funds for on-going research eorts. One way a family can make a big dierence
is to team up with an established organization or group of organizations, because
as organizations grow, they are able to make more of an impact by funding larger
research projects. Individual families and groups of families around the world
have been able to fund national and international summits for researchers. ese
forums have provided opportunities for scientists to come together to share
data and ideas. An important factor to consider when teaming up with another
organization or foundation, is whether or not the organization has a scientic
advisory board that advises the organization. is helps to ensure that the research
projects that are funded are addressing the most important issues in the eld and
are of the highest quality.
In summary, there are many ways that families can honor their children with DIPG
by helping to promote progress toward improving current treatments. It remains
clear that ongoing research is required to develop a cure for children with DIPGs.
is research requires tissue to understand the biology of DIPGs, participants in
clinical trials to evaluate new treatment approaches, increased awareness of the
devastation of this disease, and nancial support for basic science and clinical
research. Although there are hurdles and barriers related to DIPG research and
Chapter 13: Overcoming Research Hurdles in DIPG
230
treatment, they will continue to be overcome as scientists, physicians and families
work together to overcome them.

231
Chapter 14: Genomics and Proteomics in DIPG
Chapter 14
The Future of
Genomics and
Proteomics in DIPG
Mark W. Kieran, MD, PhD
e sequencing of the human genome has resulted in a revolution in biology and
medicine that oers enormous possibilities leading to an improved understanding,
diagnosis, and treatment of human diseases. ese advances have largely been
achieved in two major domains. First, the development of new technologies
that can rapidly and cheaply analyze DNA, RNA, and proteins has lead to an
explosion in our understanding of the building blocks of the cells, and how they
interact with each other through the process of growth and development. e
second major advance has occurred in the area of bioinformatics. Developments
in computational sciences have permitted the storage and analysis of the billions
of fragments of tumor data that result from these technologic advances, and
provide the opportunity to place them in the context that better approximates
complex biologic systems. us, we now have the opportunity to examine the
genome of cancer as well as begin to understand it. e goal of this chapter will
be to review the technical advances, specically genomics and proteomics, and
place these in the context of future therapeutic developments for diuse intrinsic
pontine gliomas (DIPG). ese important advances are just now being applied
to DIPG and like most other advances, will take some time before their impact
is felt in the clinical treatment of DIPG.
Genomics
In its simplest form, genomics refers to the
reading of the genetic code of cells. DNA is
the genetic material that acts as the blueprint
for making new cells as well as all of the
Dr. Mark Kieran is the Director
of Pediatric Medical Neuro-
oncology at the Dana-Farber
Cancer Institute and Children’s
Hospital, Boston, MA.
Chapter 14: Genomics and Proteomics in DIPG
232
233
Chapter 14: Genomics and Proteomics in DIPG
information needed to maintain the current ones. Tumors often alter their DNA
(the blueprint), and divide into multiple "daughter" cells which then inherit the same
altered DNA, leading to the propagation of the cancer. When a cell divides, the two
new cells formed are called daughter cells and when they divide, four daughter cells
will be created etc. Alterations in the DNA occur via changes in the genetic code
which is made up of four nitrogen bases identied by the letters: A (adenine), G
(guanine), T (thymine), and C (cytosine). ese 4 letters spell out all of the proteins
that need to be made (called the coding regions) as well as the intervening sequences of
DNA that contain the control regions (called the non-coding regions) that determine
when proteins are made. When an error occurs in the code of a cell, not only does it
have the potential to aect that cell, but that error is also transmitted to every new
daughter cell. us, errors can accumulate, increasing the malignant phenotype of
the tumor, as well as its resistance to therapy.
How do Mutations in the DNA Sequence Cause Cancer?
ere are generally two types of genes that can sustain mutations leading
to cancer: tumor suppressor genes and oncogenes. In normal cells, tumor
suppressors make proteins that keep cells "in check" (e.g. suppress tumors).
However, if errors in the DNA of tumor suppressor gene(s) are acquired, and
these errors destroy the function of the tumor suppressor, then tumors are no
longer suppressed, and the cell is not kept "in check" any longer. While tumor
suppressors cause cancer by their absence, oncogenes cause cancer by their
presence. Mutations occur to oncogenes that impart new abilities of the protein
to cause cancer. e alteration in the DNA of oncogenes results in a protein
that instructs cells to continuously divide, with uncontrolled proliferation.
e cell that possesses these types of mutations determines the kind of cancer
observed. When these mutations happen in blood cells for example, leukemia
results. If they happen in a brain cell of the pons, the patient is diagnosed
with a diuse pontine glioma. us, we consider cancer (tumor) a result of
the accumulation of abnormalities in the DNA of cells. e major types of
alterations of the DNA that can occur in pediatric brain stem gliomas (and all
other cancers) are described below.
Mutation: Genes contain the sequence code necessary to make proteins,
and the proteins make up all cells and tissues. Mutations in the DNA occur
such that the sequence (blueprint) for a protein has an error in it resulting in
defective functioning. In other words, mutations in the genetic code result in
no protein being produced, or a defective protein being made. Mutations may
involve alternations in only one letter of the code (point mutation), many letters
(nucleotides), or the entire gene (deletions, amplications, see below). If the
altered protein has a critical role in cell proliferation, then the rst step toward
development of a tumor has occurred.
Translocation: Translocations of the DNA occur when certain parts of the
DNA coding for a protein, break into two and rejoin at a site belonging to a
dierent protein. is can sometimes result in a new molecule that tells the
cell to do the wrong thing. For example, if the protein responsible for rapid
proliferation of immune cells during infection accidently gets linked to the gene
that makes astrocytes (the cells of the brain that hold everything in place), you
may create a new "molecule" that accidentally tells astrocytes to rapidly divide.
Deletions and amplications: Deleting or amplifying regions of the DNA is a
common way in which errors can be introduced into the genetic material of the
cell. If a piece of DNA coding for the molecules that stop cellular proliferation
is lost (deleted), then uncontrolled cell division can result. In a similar way, if
a segment of DNA that codes for the protein that makes cells start to divide is
amplied, then the cell is stimulated to proliferate and tumor growth ensues.
Truncation (partial deletion of a gene): Many genes are organized such that
one part possesses the functional part of the molecule (stop or start cell division)
while the other end possesses the control sequences. us, knocking out bits of
a gene, rather than the whole thing, can sometimes have a signicant impact on
the function of that molecule. For example, if the control region of a molecule
is lost, it may continue to activate the cell even at times when it should be in
the "o" position.
Epigenetic control of methylation and acetylation: Some cells are out of
control not as a result of some assault on the integrity of the DNA but because
a perfectly normal protein is expressed in the wrong place or at the wrong time.
For example, during early embryogenesis, the fertilized egg must rapidly divide
billions of times as the fetus grows. is is an example of normal proliferation. If
an astrocyte that has nished dividing accidently turns on the embryonic signal
for a cell that is supposed to be dividing, then it will begin to divide even in
the absence of any mutations, translocations, deletions, or amplications. e
process of abnormal expression or timing of a normal gene may be of particular
importance for pediatric cancers where the proliferation of many cell types
associated with growth and development are still active.
e eld of genomics is typically divided into two major components: structural
and functional. Advancements in the evaluation of the structural organization of
the DNA required technology that could rapidly, inexpensively and reproducibly
Chapter 14: Genomics and Proteomics in DIPG
234
235
Chapter 14: Genomics and Proteomics in DIPG
analyze the genetic code of both normal and abnormal tissue. is would then
allow for the identication of the mutations, translocations, amplications,
or deletions of the DNA discussed above. Improvements in this technology
are happening rapidly, and genomic analysis can now be done at both large
and small academic institutions around the world. Many of these techniques
are performed on small inert platforms (often called "chips") where millions
of reactions can be performed simultaneously. e complexity of each chip
determines how much information can be gathered from the sample being
tested. Chips are available for the study of DNA (the genetic material), RNA
(the intermediate code derived from the DNA) and proteins (the translated
end product of the RNA code into amino acids). Simply nding an alteration
in the DNA, RNA, or protein however does not prove that it is responsible
for disease. us, clinicians and scientists must map the genetic abnormalities
identied, onto the tumors ability to divide, inltrate, and escape treatment.
For example, a mutation in a protein not expressed in the tumor is unlikely
to be responsible for the tumor. us, each abnormality in the DNA, RNA,
and protein must be assessed for its active role in the tumor. Once identied,
the relevant abnormalities can then be considered for targeting with drugs or
other therapies.
Proteomics
Changes in DNA, RNA, or epigenetic events may be responsible for the abnormal
function associated with many tumors, but the analysis of the proteins themselves is
the most direct method to assess for critical changes in a cell’s function. Recall that
the purpose of DNA is to provide the code (via RNA) for all proteins. e eld of
proteomics uses a variety of techniques that allow for the separation of the thousands
of proteins in a cell, as well as the structure and function of many of these molecules.
Proteomics is usually divided into two critical phases. First is the separation of the
dierent proteins in a sample—typically achieved by their size and overall charge
(basic or acidic); and second is the identication of the dierent proteins—usually
achieved through a technique called mass spectroscopy. As with genomic analysis,
tissue is necessary for proteomics; however because many tumors shed their proteins
in the blood or cerebral spinal uid (CSF), these samples can sometimes act as
surrogates to tumor tissue. Our increasing ability to identify the location, quantity,
and activity of dierent proteins in a tumor sample has provided the pharmaceutical
industry with the targets on which tumor specic inhibitors can be developed. In
fact, a number of these drugs are already being used for a variety of dierent adult
cancers and have started testing in pediatric patients as well.
Genomics of DIPG
A primary requirement for genomic analysis of cancer, is actual tumor
material. While the biopsy of pontine gliomas was frequently performed in
the 1970s (before any of the current genomic techniques were available),
a change in national policy occurred in the 1980s for sound scientic and
clinical reasons (see chapter 19). e diagnosis of DIPG was becoming easier
to dene radiographically through CT scans in the 1970s and in particular
with MRI scans in the 1980s. Furthermore, with the very poor prognosis of
these tumors with or without biopsy, the lack of justication for a biopsy, in
which patients could experience signicant neurosurgical damage, resulted in
a moratorium on this procedure. Over the intervening 30 years, innumerable
clinical trials of radiation alone or in combination with chemotherapy,
biologic therapy, anti-angiogenic (anti blood vessel) therapy, gene therapy,
immunotherapy, etc. have been performed. None of these approaches have
signicantly altered the outcome of this disease when compared to treatment
with radiation therapy alone. Because none of these children had a biopsy, the
reason these combination therapies failed remains unknown. When biopsy
was performed, this was generally done because the tumor was atypical and
histologic assessment was needed. e majority of these atypical lesions were
discovered not to be DIPG. ese studies were important at they demonstrated
the relative safety of biopsy in this region.
Adult DIPG and animal models of pontine gliomas (not all of them are diuse
and intrinsic) are helping guide our understanding of the important genomic
changes that help maintain a tumor’s growth and resistance to therapy. Unlike
most diseases, adults rarely get DIPG and in the few reports of this disease in
these patients, the clinical course appears dierent than in children—a nding
that suggests that DIPG prefer the environment of the pediatric pons. While
it is quite easy to start tumor growth in animals for lung, breast, prostate,
or colon cancer, mice do not develop DIPG spontaneously. Fortunately, a
number of groups have been working on the development of animal models
and the rst reports of possible contenders are now available (see chapter 15).
While these models are likely to be useful in extending our understanding of
this disease, they are unlikely to provide all of the answers. As we discovered
many years ago, we have cured just about every tumor type in mice many
times over and yet those same results have often not been realized in humans.
ey may, however, become good animal models for “proof of principle
discoveries” once pediatric DIPGs are analyzed for their genomic changes.
To overcome the lack of fresh tumor material derived from the time of
Chapter 14: Genomics and Proteomics in DIPG
236
237
Chapter 14: Genomics and Proteomics in DIPG
diagnosis of patients with DIPG, many centers have initiated genomic
analysis of autopsy material. While these studies will provide some important
insight into the biology of these tumors, they all suer from the fact that the
molecular analyses performed post-mortem are altered by the initial treatment
of the tumor with the variety of procedures mentioned above (radiation,
chemotherapy etc.), and by the relatively limited sample size of these studies.
For example, in a study of 11 cases (9 autopsy and two newly diagnosed
DIPG), abnormal expression of PDGFRα in 4 cases and PARP1 in 3 cases
were identied. Equally important in these studies was the identication that
the genetic abnormalities in DIPG were dierent from malignant gliomas in
other parts of the brain. us, molecular proles of supratentorial malignant
gliomas cannot always be used to identify appropriate pathways for the
treatment of DIPG, which perhaps helps to explain the three decades of failed
clinical trials. ese studies also support a commonly held belief in the eld
that DIPGs are a heterogeneous population of tumors, and that one treatment
is not likely to be useful in all cases.
Another approach to the molecular classication of DIPG has used imaging.
Both MRI/MRS and PET/SPECT can identify individual markers in a tumor
without the need for biopsy. For example, an 11year old female with a large
pontine tumor demonstrated strong uptake of In-111-pentreotide, which
identies the presence of somatostatin receptors in the tumor. Similarly, PET
imaging allows for the detection of glucose metabolism in tumors and can
provide some important metabolic information in DIPG. As new pathways
in DIPG tumors are identied, the ability to follow tumor growth or response
with these types of imaging markers will likely make these modalities of greater
importance in the near future.
With significant advances in neurosurgical technique and previously
performed biopsies (in selected cases), the ability to safely biopsy brainstem
tumors is becoming better recognized. In a landmark study demonstrating
the safety of biopsy of DIPG, 24 consecutive children successfully underwent
this procedure in Paris. Not only did the patients not suer long-term
consequences of the biopsy, in two patients, a diagnosis other than a DIPG
was identied. e rapidity of improved neurosurgical techniques is now
opening the door to direct administration of therapy into the brainstem,
not just biopsy. As our improved molecular understanding of these tumors
continues, the ability to administer drugs directly into the pons will likely play
a greater role in treatment. A major regulator of cellular proliferation known
as p53, has been extensively evaluated in both newly diagnosed and autopsy
cases of DIPG, as well as other brainstem tumors. is critical regulatory gene
was abnormal in over half of the cases in two dierent studies. Unfortunately,
there are currently no drugs targeting p53. While only limited information
on the molecular phenotype of pontine gliomas is currently available, the
opportunity to change this is rapidly approaching.
We now nd ourselves at an important crossroads to the molecular classication
of DIPGs. For the last 30 years, it has been felt that the diagnosis of DIPG is
easily made by imaging and clinical evaluation. e risks of biopsy within the
pons were felt not to justify routine biopsy, and the moratorium on biopsy
was considered appropriate. Today, with improved neurosurgical techniques
and the availability of sophisticated genomic technologies that can derive
extensive data from very small biopsy samples, the tide has turned. DIPG
is not a single disease caused by a single mutation. Rather, there are a large
number of abnormal pathways that likely account for these tumors and only
by identifying them can we expect to develop the kinds of interventions that
will be successful.
In this regard, two exciting developments have recently been discussed at
national meetings but not yet published. e rst is the experience of the
French group that has expanded their biopsy program from 24 to 70 patients,
and has completed a prospective trial with an EGFR inhibitor in these patients.
A dierence in the outcome of children expressing abnormalities of the EGFR
pathway was signicantly better than in those who received the same therapy
but without the abnormality (suggesting their tumors were being driven by
something else). us, we may have the rst indications of an approach that
can begin to make small improvements in the time to progression of DIPGs.
While this may seem like a small step, if validated, it may represent the rst
time a therapy has really impacted the time to progression of this aggressive
disease.
e second important development is the initiation of a 20 institution
clinical trial within the United States run through the Dana-Farber Cancer
Institute, in which all patients with DIPG will undergo biopsy and extensive
molecular proling, and the treatment of these patients will be based upon
the expression of certain pathways in their tumors. us, rather than a single
treatment for everyone, each patient will receive a treatment designed for the
expression pattern of their specic tumor. From this trial, we will have the
opportunity to fully evaluate the molecular prole of newly diagnosed DIPG,
while at the same time, begin to adapt personalized therapy based on these
unique patient proles.
Chapter 14: Genomics and Proteomics in DIPG
238
e outcome for patients with DIPG remains poor. irty years of guessing
at treatment has not helped improve the outlook and has subjected countless
thousands of these children to toxic therapies that had no benet. With recent
advances in molecular technology and neurosurgical techniques, we are now
poised to investigate the underlying biology of DIPG—the rst step toward
rational and eective therapy.

239
Chapter 15: Animal Models for DIPGs
Chapter 15
Animal Models for
DIPGs
Oren J. Becher, MD
Diuse intrinsic pontine glioma (DIPG) is a rare tumor that arises in the pons of
children and is currently incurable. e only active therapeutic agent is radiation,
which unfortunately provides only temporary relief. Clinical trials for the past
30 years evaluating novel agents have failed to identify additional active and
eective agents against this tumor. In recent years, molecular genetic technologies
have been quite successful in identifying new molecular targets in numerous
cancers which has led to targeted drug development, and an increasing number
of promising targeted agents. e challenge is how to determine which novel
agents or combination of agents should move into clinical trials for DIPG. As
this is a rare tumor, it is impossible to test every new agent and combination of
these agents in these patients. ere are not enough patients to accomplish this
feat. An additional complexity to new drug development is that DIPG tumors
are heterogeneous and may be comprised of multiple dierent subtypes, where
each subtype may respond dierently to specic drugs.
Why Do We Need Animal Models?
One idea that scientists thought might be helpful is to develop animal models that
would allow for screening of novel agents and combinations. e results from these
animal models could be predictive of anti-tumor activity in children with DIPGs,
leading to the discovery of the most promising drugs for human DIPG trials. At
this point in time, this ideal has yet to be fully realized. ere is currently one main
obstacle that must be overcome so that
predictive animal models can be developed.
Scientists need a better understanding of the
genomic alterations that drive the growth of
human DIPGs so as to guide the development
of accurate animal models to potentially treat
Dr. Becher is an Assistant
Professor in the Departments
of Pediatrics and Pathology
at Duke University School of
Medicine, Durham, NC.
Chapter 15: Animal Models for DIPGs
240
241
Chapter 15: Animal Models for DIPGs
it. Unfortunately, scientists do not have a good understanding of the drivers of these
tumors, although there are several research groups who are currently working on this,
and it is believed that answers to these questions are within reach.
In spite of the limited understanding of the biology of human DIPGs, there are several
animal models that have been described, although it is not clearly known if they are
predictive. In the next few paragraphs, I will review the various DIPG models that
currently exist, and the advantages and disadvantages of each.
DIPG Animal Models Available
Most of the current animal models for DIPGs are rat allograft models and only
recently a genetically engineered DIPG mouse model was developed as well.
e three main types of animal models for glioma are:
1. Allograft: Chemically induced tumors in rats that mimic DIPG.
2. Xenograft:
Human tumor from patients, transplanted into mice or rats.
3. Genetically Engineered Mouse Model/GEMM: Mice given new genes
(transgenics) that cause tumors appearing similar to DIPG, and sharing
some molecular signatures of the human tumor.
Allograft: ere are several rat glioma cell lines available, which were generated
by injecting rats with repeated dosing of chemotherapy until the rats developed
gliomas, which were then cultured and propogated. ere are currently several
rat glioma cell lines available. ree of these cells lines (C6, 9L, and T9 gliomas)
were induced by repeated injections of methylnitrosurea (MNU) to adult
rats. Two other cell lines (RG2 and F98 gliomas) were chemically induced by
administering ethylnitrosurea (ENU) to pregnant rats. In this case, the progeny
developed brain tumors that subsequently were propagated in vitro and cloned.
Both MNU and ENU are alkylating agents, which mean that they damage
DNA by adding alkyl groups to it. Most of the above mentioned rat glioma
cell lines are being used to generate brainstem gliomas by direct injection
into the brainstem of either rats of the same strain or immunodecient rats.
Depending on the number of cells injected, the cell line used, and the age of
the rats at the time of injection, the rats go on to develop brainstem tumors one
to several weeks later. A head-to-head comparison between 3 week-old and 10
week-old rats injected with the same cell line and the same number of cells into
the brainstem demonstrated that the microenvironment of young rats allows
for the formation of diuse pontine tumors, while the microenvironment of
older rats allows for the development of focal brainstem tumors. Interestingly,
there have not been many trials testing systemic chemotherapy using such
models. is is most likely due to the belief that systemic chemotherapy for
the most part does not get into the brain tumor due to the blood-brain barrier
(BBB). erefore, most of these models have been mainly used to test for CED
(convection enhanced delivery) of chemotherapy such as carboplatin. A detailed
description of CED is given in chapter 17 of this book.
Xenograft: Recently, a rat xenograft model was developed whereby adult
human glioma cell lines were implanted into 6-week old immunodecient
rats. Prior to implantation, some of the cell lines were maintained in media
with serum (these are usually grown as cells adherent to plastic); some of the
cell lines were maintained as subcutaneous xenografts (grown under the skin
of immunodecient rats); and one cell line—the GS2 cell line was maintained
as neurospheres (these are spherical colonies in suspension in media).
It is well documented that gliomas which are cultured in epidermal growth
factor (EGF) and basic broblast growth factor (bFGF), and are grown as
neurospheres, have genomic signatures that most resemble the signatures of
naturally developing (in vivo) gliomas. While an advantage of such a model is
that the tumor cells are human, the disadvantage is that adult glioma cell lines
have dierent genetic alterations than DIPGs. In addition, the in vitro culturing
step likely alters the biology of the cells even in neurosphere conditions. Lastly,
such models remove the role of the immune system in DIPG tumorigenesis.
GEMM: Genetically engineered mouse models for brain tumors have been
developed since 2000 and are a more recent addition to the animal modeling
toolbox. e advantage of such models is that the genetic alterations which
initiate and drive tumor formation are known and recapitulate the genetic
alterations present in the respective human tumors. erefore, the genetic
alterations of the respective human tumors should be determined so as to guide
the development of the genetically engineered mouse model. Such models
are helpful in determining if a particular genetic alteration can drive tumor
formation. Not all genetic alterations are equally meaningful and scientists divide
genetic alterations into “drivers” and “passengers” to imply that only certain
genetic alterations can drive tumor growth (so-called driver mutations) while
the role of other genetic alterations is less clear (so-called passenger mutations).
ere are numerous technologies that can be used to generate these mice. One
brain tumor model uses conditional knockout mice where mice that have
lost one copy of p53, PTEN, and NF-1 develop brain tumors through loss
Chapter 15: Animal Models for DIPGs
242
243
Chapter 15: Animal Models for DIPGs
of heterozygosity. A second system is the RCAS-tv-a system, which allows for
oncogene delivery by injection of virus producing cells into areas of interest such
as the brainstem. Normal mice do not express the receptor (called tv-a) and so
are not susceptible to infection by RCAS vectors. However, two transgenic mice
were developed that express the tv-a receptor in progenitor/stem cell of the brain
compartment: nestin tv-a mouse and GFAP tv-a mouse. (A transgenic mouse
is a mouse which integrates an additional piece of DNA in its germline called
a transgene, and so every cell in the mouse acquires this extra piece of DNA.)
Recently a genetically engineered mouse model for brainstem gliomas was
developed which recapitulates the genetic alterations of a subset of the human
disease. It was recently observed that PDGFRα is amplied in 20% to 30%
of DIPGs which means that the receptor for PDGF ligand is expressed in
high levels in a subset of DIPGs. e derived mouse model uses the retroviral
delivery system described above, whereby a virus is used to deliver oncogenes to
specic areas in the mouse brain. Tumors are generated by the over-expression
of PDGF-b in nestin positive cells which line the oor of the 4th ventricle
at postnatal day 1 to 3. Nestin positive cells are cells that express nestin,
an intermediate lament that is expressed in progenitor cells in the brain.
Overexpression of PDGF-b results in the formation of low-grade brainstem
gliomas. e addition of Ink4a-ARF null genetic alteration (which has been
described as a common alteration in human DIPGs and normally restrains
cell division), together with PDGF-b overexpression, results in the formation
of high-grade brainstem gliomas, or DIPGs, with high incidence by six weeks
of age.
Advantages of genetically engineered mouse models are that a) the genetic
alterations are clearly dened; b) the tumor forms in the right microenvironment
(the brainstem); and c) the tumor forms at the right time period (pediatric). It
is generated completely in vivo and develops de novo in the mouse. As tumors
are a complex cellular setting, it is important that this environment is as close
to reality as possible. Another advantage of the genetically engineered mouse
model is that it can be used to determine the cells-of-origin for a particular
tumor. e cells-of-origin for the recently developed DIPG mouse model were
derived from cells lining the oor of the 4th ventricle and aqueduct. However it
does not tell us with any certainty that the cells-of-origin for human DIPGs are
similar cells. One disadvantage of this animal model is that it is probable that this
genetically engineered DIPG model may be oversimplied, as human DIPGs
likely contain more genetic alterations than simply PDGF-b overexpression
and Ink4a-ARF loss. It remains to be determined whether therapeutic agents
with antitumor activity in this animal model will also be active in children
with DIPGs.
ere is no perfect animal model. Ultimately there is a need for a predictive
model of activity in the clinic. An added complexity is that the human tumors
are heterogeneous and so it is likely that we will need to classify them into
several groups based on their genetic alterations. Each subtype will then require
specic therapy and an associated specic DIPG animal model. Of note, adult
gliomas have recently been subdivided into three groups based on the genetic
alterations of the tumors.
One advantage of rat brainstem glioma models is that the rat brainstem is
larger than the mouse brainstem. As a result, it may be easier to conduct CED
preclinical studies. e disadvantage of rat allograft models are that in most cases
the genetic alterations of the tumors are not clearly dened and may change
over time. Most of the cell lines are maintained on plastic dishes that are quite
articial. In addition, as mentioned, human tumors are complex with several
cell types interacting including tumor cells and various stromal cells such as
blood vessel cells, support cells, and immune cells. erefore it is ideal for the
animal to develop the tumor within a normal immune system and with all of
the support cells being present from tumor onset.
Preclinical Testing
e question that often arises, is how much preclinical evidence is needed before a
decision is made to move a new agent into the clinic for DIPGs? ere are several
levels of preclinical evidence and I personally believe that novel agents should move
to the clinic after full preclinical testing has been done. is means that a novel
agent has been tested in cell lines; in DIPG xenograft rat models where the xenograft
originated from a DIPG tumor that has been propagated in vivo or, second best,
neurospheres; and has also been tested in genetically engineered DIPG subsets.
Depending on the therapeutic agent, some agents cannot be tested in cell lines at all
and can only be tested in vivo or in neurospheres. An example is the sonic hedgehog
pathway inhibitors, which cannot be tested in cell lines grown on plastic dishes, as
the pathway is not functional in such conditions.
Once an agent shows strong promise in preclinical testing, a decision is then made
to test it in patients with DIPGs. e rst clinical study is a phase I study which is
used to determine the correct dose to use in the clinic, as well as assess for toxicities
of the drug. Even if a drug has already been tested in adults, it will still need to be
tested in a phase I trial for children as children at times metabolize drugs dierently
Chapter 15: Animal Models for DIPGs
244
than adults. Usually phase I studies are open for children with diverse cancers but it
does not mean that a particular drug will not be active in DIPGs. Once the safety and
dosing are established in a phase I study, then a phase II trial is designed to determine
if the drug or drug combination is active against DIPGs. Phase II studies are usually
done on a selected tumor subtype. Phase III studies are large studies that are used to
conrm promising results from phase II studies (see chapter 5).
Conclusion
At this point in time, most clinicians do not believe that animal models can help
guide which therapeutic agents or combination of agents will be active in the clinic.
e burden of proof lies with the animal-modeling eld to continue to improve
the animal models so that eventually they will be predictive of activity in the clinic.
Similar to what has been done in adult gliomas, in the near future DIPGs will also
be grouped into genomic subgroups, and genetically engineered animal models will
be developed for each subtype. It is my hope that these genetically engineered DIPG
mouse models will be predictive of activity in the clinic, but it remains to be seen if
this will indeed be the case.

245
Chapter 16: Neural Stem Cells and DIPG
Chapter 16
Neural Stem Cells and
DIPG
Michelle Monje, MD, PhD
Diuse intrinsic pontine gliomas (DIPG) occur strictly in the ventral pons
and typically during a relatively specic period during mid-childhood, peaking
between ages 6 and 8. e age and location-specic nature of DIPG suggests that
the underlying pathophysiology may involve dysregulation of a developmental
process. In this context, it makes sense to approach DIPG from the vantage of
neural stem and precursor cell biology.
Normal Neural Stem Cells
Neural stem cells—cells that can renew themselves and also can make all types of
neural cells (neurons, oligodendrocytes and astrocytes), are well-recognized in the
brain and spinal cord of both children and adults. Two populations of neural stem
cells are very well studied. ese two populations reside in the hippocampus, a brain
structure important in memory function, and in what is called the subventricular
zone (i.e. just below the ventricular walls) of the lateral ventricles. It is known that, at
least in mice and rats, stem cells exist throughout the ventricular system of the brain
and spinal cord, but little attention has been paid to those in the subventricular zone
of the third and fourth ventricles. e fourth ventricle sits immediately behind the
pons. e term “neural precursor cell” includes both true stem cells and cells that
are somewhat further along the path of dierentiation but still give rise to daughter
cells. Both types of cell—stem and “precursor”
are important to developmental processes in
the brain both before and after birth.
Cancer Stem Cells
Cancer stem cells (CSCs) represent a
subpopulation of cells that can generate all
Dr. Monje is an Assistant
Professor of Neurology and
Neurological Sciences at
Stanford School of Medicine,
and Practicing Pediatric Neuro-
oncologist at Stanford Hospital,
and Lucile Packard Children’s
Hospital, Stanford, CA.
Chapter 16: Neural Stem Cells and DIPG
246
247
Chapter 16: Neural Stem Cells and DIPG
cell types found within a tumor and are thought to be responsible for tumor
growth and spread. Like normal stem cells, cancer stem cells possess the capacity
for self-renewal and multi-potency. (Multi-potent cells can make all the cell
types in a tissue. In this case the tissue is the tumor.) e rst cancer stem
cells were described in acute myeloid leukemia, and have now been shown in
many solid tumors, including many brain tumors such as glioblastoma and
ependymoma. CSCs isolated from primary brain tumors possess many of the
characteristics of normal neural stem cells, and can recapitulate the tumor in
vitro and in vivo, whereas other cell types from the tumor cannot. CSCs are thus
a small proportion of a tumor, but are solely responsible for tumor propagation.
e relationship of normal neural stem cells to cancer stem cells is somewhat
controversial, but there is an emerging consensus that many brain tumors
arise from stem or precursor cell populations in both children and adults.
Excellent examples of this point include “radial glia” cells (a type of stem cell)
giving rise to ependymoma and subventricular zone neural stem cells giving rise
to central neurocytomas. With respect to more lineage-restricted precursors,
Shh-responsive granule cell precursor cells of the cerebellum give rise to
medulloblastoma in many cases, and recent animal model data indicate that
oligodendrocyte precursors give rise to periventricular low grade gliomas in a
mouse model of platelet-derived growth factor (PDGF) overexpression. Brain
tumor stem cells exhibit many of the same marker proteins and utilize many
of the same signaling pathways as normal neural stem cells. Understanding
normal neural stem or precursor cells in the brainstem may thus shed light on
brainstem tumor pathogenesis.
What Stem Cells Need to rive—the “Stem Cell Niche”
Normal stem cell niche
Neural stem cells in the childhood and adult nervous system reside in a niche
of signaling factors, extracellular matrix composition and specialized cell types
that support neural stem cell function for that brain region. Perhaps best studied
is the stem cell niche that supports forebrain neurogenesis in the hippocampus.
is specic microenvironment necessary for stem cell production of new
neurons is referred to as the neurogenic niche. Transplantation experiments
demonstrate that neurogenesis is restricted in the postnatal brain to regions
in which it occurs naturally, namely the subventricular zone (SVZ) and the
subgranular zone (SGZ) of the hippocampus. In general, microenvironmental
determinants of neurogenesis include the presence of the trophic signals
required for progenitor cell proliferation, dierentiation and survival, and
the absence of inhibitory factors. Neural stem/precursor cells form a close
anatomical relationship with the small vessels in the neurogenic region, and this
neurovascular relationship—the so-called “vascular niche”—is believed to be
crucial not only for nutritional but also for growth factor support. Vessel cells
(endothelial cells, pericytes) and glial cells (astrocytes) all contribute to the stem
cell niche. Hippocampal astrocytes play key roles in creating and maintaining
the neurogenic niche. As noted above, many of the signaling pathways central to
prenatal neural development are conserved in postnatal neurogenesis, including
pathways called Wnt, Shh, and Notch. Additional molecules with potent pro-
neurogenic eects include broblast growth factor (FGF), vascular endothelial
growth factor (VEGF) and certain neurotransmitters. An important negative
regulator of the neurogenic microenvironment is microglial cell inammation,
particularly in disease states. Pro-inflammatory cytokines elaborated by
microglial cells in certain states of activation, including IL-6 and TNF-alpha,
inhibit neurogenesis via a specic blockade in neuronal dierentiation mediated
by Notch signaling, as well as a non-specic increase in precursor cell death.
e eects of inammatory cells on neurogenesis are complex and depend on
the microglial phenotype involved; microglia stimulated by cranial irradiation
or systemically-administered lipopolysaccaride (LPS, also known as endotoxin)
inhibit neurogenesis, while microglia stimulated by IL-4 or interferon gamma
promote neurogenesis.
Cancer stem cell (CSC) niche
Just as cancer stem cells share many properties with normal stem cells, so
the cancer stem cell niche is similar to the normal stem cell niche. e
vascular niche appears to be recapitulated in human brain tumors. Cancer
stem cells, dened molecularly by expression of the proteins CD133 and
Nestin, are localized in close proximity with tumor microvessels in human
medulloblastoma, glioblastoma, oligodendroglioma and ependymoma. e
relationship between cancer stem cells and tumor microvessels is bidirectional:
glioblastoma cells induce angiogenesis (new vessel cell growth) via VEGF
elaboration, and vascular endothelial cells supports glioblastoma cell
tumoriogenicity. Treatment of a mouse orthotopic glioblastoma model with
the VEGF blocking agent bevacizumab (Avastin) depletes CD133+ cells,
decreases tumor vascularity and reduces tumor growth rate. Accordingly,
bevacizumab has shown modest clinical ecacy in glioblastoma, at least in
adult glioblastoma of the forebrain. Highlighting the dierences between
DIPG and adult glioblastoma, bevacizumab is not ecacious for DIPG.
Important determinants of the DIPG cancer stem cell niche are yet to be
dened.
Chapter 16: Neural Stem Cells and DIPG
248
Stem Cells in DIPG
Cell of origin
Presently, intense research is underway to identify the cell type in the normal
childhood pons from which DIPGs originate. e cell type that transforms
and gives rise to DIPG could be a neural stem cell, a neural precursor cell type
(that is destined to give rise to glial cells or neuronal cells) or a dierentiated
cell type (glia or neurons). Lessons learned from other pediatric brain tumors
teach us that the most likely candidate would be a neural stem or precursor
cell. Neural stem and precursor cells are not well described in the brainstem,
but current research will soon shed light on candidate cells of origin for DIPG.
Understanding the cell of origin for DIPG is of fundamental importance to
elucidate mechanisms by which DIPG may form, and thus potential targets
for treatment.
DIPG cancer stem cell
Researchers are working to identify and characterize a cancer stem cell in DIPG.
is research requires fresh tumor samples for cell culture, and scarcity of tissue
for research has limited progress in this area until very recently. Donation
of tumor in the early post-mortem period after the loss of a child allows for
successful cell culture of both normal brain and brain tumor tissue. is
strategy can allow crucial research to be done without putting a child through
an additional procedure such as a biopsy. Identifying and studying the cancer
stem cell of DIPG may elucidate new targets for therapy that are at the core of
DIPG growth and propagation.
A few words on hematopoietic stem cell and bone marrow transplant
Hematopoietic stem cell transplant (HSCT) and bone marrow transplant are
designed to rescue the bone marrow after intensive chemotherapy or to provide
cell replacement therapy for certain genetic diseases. At present, there is no role
for either HSCT or bone marrow transplant for DIPG.

249
Chapter 17: Convection-Enhanced Delivery in DIPG
Chapter 17
Convection-Enhanced
Delivery in DIPG
Zhiping Zhou, MD, PhD
Mark M. Souweidane, MD
Diuse intrinsic pontine glioma (DIPG) is locally highly inltrative. In this
type of tumor, cancer tissue cannot be distinguished from normal brain tissue
macroscopically. is inltrative nature makes eective therapy extremely dicult,
if not impossible. To be clinically useful, a therapy must have the ability to
selectively target and kill tumor cells without signicant damage to the normal
brain tissue. Given the need for cell specicity, surgical resection and stereotactic
radiosurgery have limited utility, pose a great risk of injury and are therefore rarely
an option to consider. Conventional radiation therapy is currently employed
routinely as a palliative approach. e improvement of newer chemotherapeutic
agents’ tumor selectivity and the development of targeted therapeutic agents in
recent decades raise hopes that improved chemotherapy will lead to improved
outcome. Paralleling this development has been the promising advance in drug
delivery to the central nervous system via local delivery systems to overcome the
blood-brain barrier (BBB), one of the major hurdles in delivering drugs to the
brain.
In this chapter, we will be discussing the application of convection-enhanced
delivery (CED), also known as interstitial infusion, in the treatment of DIPG.
CED is a technique designed to deliver drugs directly into the tumor at high
concentrations. is avoids or at least greatly reduces systemic exposure to the
drug. Drugs being studied for delivery through CED include conventional
chemotherapy drugs, novel small molecule agents and macromolecules such as
therapeutic antibodies, immunotoxins, and
viral vectors, some of which would otherwise
never gain access to the brain.
Dr. Zhou is an Instructor in the
Department of Neurological
Surgery at Weill Cornell Medical
College, New York, NY.

Chapter 17: Convection-Enhanced Delivery in DIPG
250
251
Chapter 17: Convection-Enhanced Delivery in DIPG
Blood-Brain Barrier
Chemotherapy may be administered systemically or locally. In systemic chemotherapy,
the drug is administered orally or intravenously. An important limitation of systemic
chemotherapy in the treatment of brain tumors is the existence of the blood-brain
barrier (BBB). e BBB isolates the circulating blood from cerebrospinal uid
(CSF) in the central nervous system (CNS). It occurs along cerebral capillaries and
consists of tight junctions (zona occludens) that do not exist in systemic circulation.
Endothelial cells joined by tight junctions restrict the entry of microscopic objects
(e.g., bacteria) and large or hydrophilic molecules into the CSF, while allowing
the diusion of small hydrophobic molecules (O
2
, certain hormones, CO
2
, etc.).
Typically, molecules with molecular weight greater than approximately 40kD are
unlikely to penetrate the intact barrier. For the brain’s supply of nutrients and removal
of metabolites, cells of the barrier actively transport substances such as glucose across
the barrier with specic proteins (transporters). e BBB acts eectively to protect
the brain from many common bacterial infections and some toxic substances. Yet
it presents a major challenge in delivering therapeutic agents to specic regions of
the brain for the treatment of brain tumors and other disorders. Most cancer drugs
are not able to permeate the BBB because they are polar in structure or too large in
molecular weight. Even for drugs that are able to cross the cerebral capillary bed, it
is dicult to achieve optimal concentrations due to systemic toxicity.
Another diculty in the delivery of drugs for the treatment of brain tumors is how
to direct those agents to the specic anatomic region or tumor mass to reduce the
disturbance of normal neurological functions. Several strategies have been developed
in an attempt to overcome this barrier, including: 1) the temporary disruption of
the BBB, 2) modication of drugs to enhance their ability to permeate the BBB and
3) local delivery methods such as intratumoral/intra-cavitary embedding of drug-
containing polymers or microchips, intra-arterial injection, direct injection of drugs
into the tissue or CSF in the ventricles or subarachnoid space, and CED to deliver
drugs directly into the extracellular space.
Local Delivery
Direct injection into the tumor or CSF is
one of the earliest local delivery methods
attempted. When injected into the tumor,
it relies on diusion for the drug to reach
the cancer cells not directly adjacent to the
injection site. erefore the drug has an
uneven distribution and can only reach the
tumor tissue that is a short distance from the injection site. With small molecules,
depth of distribution is often limited to several millimeters, with an exponential
decay in concentration from the point source. us, the distribution of therapeutic
concentrations of a drug is limited to a small volume of tissue around the injection
site, often with very high and sometimes toxic concentration at the center. Drugs
can also be injected directly into the CSF, and the drug is usually only able to
reach a shallow layer of the brain using this technique.
Drug-containing polymers and microchips are a more recent development and
they can be embedded at the time of surgical resection of brain tumors. As in the
case of direct injection, this method relies on diusion for the drug to spread past
the embedding site and has similar limited and uneven distribution.
Convection-Enhanced Delivery
Convection-enhanced delivery (CED) is a novel drug delivery method rst
developed by a research group directed by Edward Oldeld at the National
Institute of Neurological Disorders (NINDS) in the early 1990s. is method
was named convection-enhanced delivery because the therapeutic molecules are
distributed into the extracellular space driven by a small, persistent hydrostatic
pressure generated by an infusion pump, essentially, forced convection of a uid
containing a therapeutic agent. In contrast to diusion which depends on a
concentration gradient to distribute the molecules, the use of hydrostatic pressure
in CED allows for the distribution of a homogeneous concentration of small
and large molecules over large distances by displacing extracellular uid with the
infusate (uid infused). In practice, the agent is delivered into the parenchyma
or tumor through a microcatheter, or multiple microcatheters, inserted into the
tissue. Infusion rates typically range from 0.1-10µl/min. e distribution from
a single point source results in an elliptical to spherical distribution and spatial
distribution is in some degree dependent on the tissue type (i.e., grey matter
versus white matter). In a given tissue type, distribution volume is approximately
linear to infusion volume.
CED into brain parenchyma, both white and gray matter, has shown reproducible
large volumes of distribution with homogeneous drug concentration. Oldeld
group’s initial work showed that the concentration fall-o at the border is steep,
resulting in a potentially large benet in the delivery of cancer drugs in reducing
toxicity to surrounding normal brain tissue.
Several factors inuence the distribution volume. One key factor to achieve a
large volume of distribution is the stability of the agent in the extracellular space.
Dr. Souweidane is the Director of
Pediatric Neurological Surgery
at Weill Cornell Medical College
and Memorial Sloan-Kettering
Cancer Center. He is also
Vice Chairman and Professor
of Neurological Surgery and
Professor of Neurological
Surgery in Pediatrics at Weill
Cornell Medical College, New
York, NY.
Chapter 17: Convection-Enhanced Delivery in DIPG
252
253
Chapter 17: Convection-Enhanced Delivery in DIPG
Lipophilic agents may be exported transvascularly through blood vessels leading
to a high eux of the drug and limited distribution. Some other drugs may be
prone to enzymatic degradation in the extracellular space. Another important
determinant for distribution of macromolecules is the surface characteristics of the
molecule and the extracellular matrix, i.e., the substances in the extracellular space
within tissues that serve various purposes, including but not limited to serving as
scaolding to hold tissues together and helping to determine the behaviors of the
cells. Binding of the molecule to the extracellular matrix or surface receptors may
limit distribution. Binding to cell surface receptors may be overcome by saturating
receptors with excess ligands. Binding of macromolecules to extracellular matrix
has been overcome with some success by co-infusion of heparin.
Size of the molecules also aects volume of distribution. Early CED studies by
Oldeld group and others suggested that 180kD, the size of immunoglobulin
G (IgG), appeared to be the largest size that could pass through the extracellular
space without the need of surface modication to the extracellular matrix.
Recently, with the help of surface modication, adeno-associated virus (AAV,
40nm) and liposomes (50-200nm) have been distributed to large volumes of brain
tissue. Surface modications used were pegylation with liposomes and heparin
co-infusion to saturate heparin sulfate proteoglycan (HSP) binding with AAV.
e volume of distribution is also aected by the retrograde movement of uid
along the outside of the catheter (backow or reux). Reux is determined by
catheter diameter, infusion rate, and tissue density among other factors. e
larger the diameter of the catheter, the greater is the backow along its outer
wall. If reux reaches a low pressure zone (necrosis or CSF space), the uid will
inadvertently be lost into these spaces. is leads to the accumulation of the drug
in these regions which may cause toxicity. Finally, increasing the infusion rate
can increase the overall volume of distribution; however, this may also increase
backow, potentially shunting uid away from the target region.
Ideally, agents delivered via CED should be contained within the target region of
brain parenchyma or tumor mass. However, there are low pressure regions in some
tumors along which infusate will ow, sometimes into ventricles or subarachnoid
space. is phenomenon is usually referred to as leakage and has often been
observed in both humans and experimental animals. One study indicates that this
can happen in 20% of CED procedures. is obvious waste of therapeutic agents
will consequently reduce the volume of distribution and drug concentration in
the planned target region. It may also cause untoward eects on normal brain
tissue. It is therefore critical to follow the ow of the infused agents. When this
happens, it might be helpful to adjust the placement of the catheter to move the
opening away from the low pressure region. It is also not known yet whether
this leakage is reversible. If reversible, pausing infusion for a period of time and
subsequently restarting the infusion could eliminate leakage.
Although the physical parameters inuencing drug distribution by CED have
not been thoroughly claried, the ability of CED to achieve high concentrations
of a therapeutic agent over large volumes of brain tissue has led to several clinical
trials in patients with neurodegenerative disorders and malignant gliomas.
erapeutic studies for malignant gliomas have focused on delivering targeted
macromolecules (monoclonal antibodies, recombinant toxins, etc.) or currently
available small molecule drugs.
Catheter Design for CED
Metal needles have been used as the infusion tool since the early studies of CED
in laboratory animals. Most of the recent clinical trials of CED in the treatment
of malignant gliomas have used ventricular catheters made of Silastic® rubber.
Ideally, a catheter for CED should be reux-free; does not adsorb therapeutic
agents to its wall, especially when expensive novel targeted agents are used; and
should have tip congurations that direct the drug to desired regions. In certain
instances, it may be required to conrm catheter placement before drug infusion
with magnetic resonance imaging (MRI) where MRI-compatible catheters are
needed.
As briey discussed above, reux negates the bulk ow of infusate in the
extracellular space that is produced by CED. In the presence of reux, an
increase in infusion volume does not produce an increase in distribution volume
accordingly. Reux causes the drug to ow into ventricular or subarachnoid
space where it may cause toxicity. While reduction in infusion rate may reduce
the chance of reux, it would be ideal to have the option of infusing at various
ow rates, i.e., up to 10µl/minute or more if possible, to achieve desired volume
of distribution in a reasonable period of time.
Simple infusion tools such as metal needles have high rates of reux. Several
groups, including Souweidane group at Weill Cornell Medical College, observed
that a step-design cannula signicantly reduces, or even eectively prevents,
backow. e group used a 22 gauge guide cannula with a 28 gauge internal
cannula, both of fused silica. e internal cannula extended beyond the end of
the guide cannula by 5 mm. e cannula set was left in place for 5 minutes before
infusion started. At ow rates as high as 8µl/minute of an
124
I-labeled monoclonal
antibody, no reux was observed on positron emission tomography (PET)
Chapter 17: Convection-Enhanced Delivery in DIPG
254
255
Chapter 17: Convection-Enhanced Delivery in DIPG
imaging. Presumably the tissue surrounding the extended internal cannula sealed
o the entry tract. ere might be a threshold that this design can withstand the
pressure, but infusion rates higher than 8µl/min have not been attempted with
this tip conguration. Nevertheless this design oers an attractive improvement
over the cannula design previously used. is fused silica cannula set is not and
probably will never be approved for clinical use due to its insucient mechanical
strength. Ventricular catheters currently used in CED clinical trials have larger
diameters and would produce a higher chance of reux based on laboratory
observation of the relationship between reux and cannula diameters. Advances
in biocompatible materials such as polymers and ceramic may eventually make
small diameter MRI-compatible step-design cannulas and catheters strong enough
for clinical use.
Tip Conguration
A standard cannula only has an opening at its tip. In certain instances, such as
after radiation therapy where scars may form inside the tumor, this may not
allow for sucient ow of infusate. Considering infusates will follow the path
of least resistance, a multi-tipped cannula may provide better pressure output,
and therefore, achieve a better volume of distribution. e eectiveness of the
multi-opening conguration has been questioned by studies showing that a
multi-port catheter delivered most of the infusate through the proximal port
and thus behaved like catheters with only one port.
One research group constructed a 3-mm long porous hollow ber catheter
to increase the surface area of the brain in immediate contact with the drug
releasing area. e hollow ber has innumerous pores of 0.45µm along its
walls. is theoretically avoids clogging, which happens in certain instances.
e hollow ber catheter oers up to a threefold increase in the distribution
volume of the drug into the normal mouse brain when compared to a needle
which has a single macroscopic pore. e tiny microscopic pores do not have
the same pressure-shunting properties as the macroscopic pores do; therefore
a long length of the porous wall is eective in delivering drugs. In large animal
and human applications, it is more reasonable to have this porous hollow ber
conguration at the tip for a few millimeters to a few centimeters rather than
the entire catheter being porous. e porous wall and step design could even
be combined to reduce reux during drug administration.
In certain other instances, it may be desirable to direct the infusate preferentially
in a specic direction. Due to the pressure-shunting properties of the proximal
port on the regular multi-port cannulas, it may not be eective to direct infusate
distribution via such a tip conguration. One potential design is to construct
a catheter with independent cannulas inside. Each cannula has an opening at
a predetermined location and direction with its pressure being independently
controlled. is design will require additional engineering and testing to
determine its feasibility.
Monitoring Drug Distribution
Monitoring the distribution and concentration of an infused drug is critical for
numerous reasons. In order for the delivered therapeutic agent to be eective, in
addition to its biological eectiveness, it must be distributed within the tumor
in therapeutic concentrations. Exposure of normal tissue to the drug should
be controlled to reduce the probability of toxicity. It is also highly desirable
to monitor for possible backow and leakage so that cannula placement
can be adjusted to correct for any problems that may arise. e importance
of monitoring in vivo distribution and concentration is highlighted by the
diculty in achieving optimal therapeutic ecacy in recent clinical trials. In
the recent TGFα-PE38 study and the phase III PRECISE trial for glioblastoma
(see below), poor drug distribution was cited as one of the reasons for the
unsatisfactory ecacy results. Monitoring the distribution and concentration
of CED infusate in humans is dicult due to the fact that the majority of
therapeutic agents cannot be seen on any of the clinical imaging methods.
Nevertheless, distribution can be visualized under certain circumstances. T2-
weighted magnetic resonance (MR) images are helpful in identifying infusate
distribution in regions of relatively normal intensity, but distribution cannot
be identied with certainty when infused into already hyperintense regions,
such as in the case of DIPG.
Another choice is to use visible surrogate tracers. Gd-DTPA and
123
I-albumin
have been co-infused as surrogate tracers, viewable on T1-weighted MR and
single photon emission computed tomography (SPECT) images, respectively,
in clinical studies. e shortcomings of surrogate markers are that they are
only able to track the initial distribution accurately. Dierences in biological
activities and clearance can confound their ability to follow the volumetric
distribution of the therapeutic agent over time. Moreover, neither T2 MRI
signals nor surrogate tracers are able to provide information on the concentration
of the infused therapeutic agent. e ideal scenario is to directly image the
therapeutic compound. With calibration, the concentration of the drug can be
determined as well as the distribution. Utilizing serial imaging, clearance can
be followed over time. In an ongoing clinical trial at Memorial Sloan-Kettering
Chapter 17: Convection-Enhanced Delivery in DIPG
256
257
Chapter 17: Convection-Enhanced Delivery in DIPG
Cancer Center and Weill Cornell Medical College, a therapeutic monoclonal
antibody is labeled with
124
I to treat DIPG.
124
I is a positron emitter that can
be visualized using PET imaging at a high resolution. e spatial resolution of
124
I PET is signicantly higher than that of
123
I SPECT.
124
I has an intrinsic
spatial resolution loss of only 2.3 mm. It is expected that much more detailed
information regarding the distribution and concentration of CED infusate
will be acquired. is approach of labeling a therapeutic agent with imageable
radionuclide can be applied to some other agents and applications. For some
other therapeutic agents, novel tags such as paramagnetic particles may prove
useful in labeling the drug for quantitative in vivo imaging.
Predicting and Planning CED Distribution
It is critical to dene the relationship between the volume of infusion (Vi) and
the volume of distribution (Vd) to understand the expected distribution of an
agent delivered into the brain via CED. is relationship is approximately linear
and has variable slopes depending on the anatomical site of administration as
well as the therapeutic compound. For instance, the Vd/Vi ratio is 8.2 in the
non-human primate (NHP) striatum compared to a ratio of 4.1 in cerebral white
matter for small molecules. A ratio of 8.7 was observed in the NHP brainstem
for Gd-albumin (72kD). is ratio can serve as an estimate to match tumor
volume in clinical trials.
BrainLAB AG (Feldkirchen, Germany) has developed a software package
called iPlan Flow specically for use in planning CED. e software takes data
obtained via MRI regarding brain tissue characteristics of individual patients as
input. en the software helps in determining cannula placement, calculating
the infusion parameters and predicting distribution. e plan for treatment
can be visualized in three dimensions, including the number and position of
catheters. One study retrospectively tested the ability of this software using
MR diusion tensor imaging to predict patient-specic drug distributions by
CED.
123
I-labeled albumin was co-infused as a surrogate tracer with the targeted
recombinant cytotoxin IL13-PE38QQR in patients with recurrent malignant
gliomas. e spatial distribution of
123
I-albumin was then compared with a
drug distribution simulation provided by iPlan Flow. e algorithm had a
high sensitivity and specicity in identifying catheter trajectories that resulted
in reux or leakage. e mean concordance of the volume of distribution
between the actual
123
I-albumin distribution and the simulation was 66% and
the mean maximal inplane deviation was less than 8.5 mm. e use of this
simulation algorithm was considered clinically useful in 85% of the catheters.
Even though albumin does not have a specic anity towards malignant tissue
compared to targeted agents, this simulation showed that software with the
ability to take into account characteristics of an individual patient’s anatomy
and pathophysiology is helpful in the planning of CED. iPlan Flow has yet to
be tested in CED in the brainstem.
Safety of CED in the Brainstem
e concept of using CED for DIPG is appealing given that this particular
tumor is relatively compact, has growth patterns simulating white matter
tracts, seldom metastasizes before local relapse and has no denitive therapy.
e Souweidane group rst established the feasibility of this delivery route
in the brainstem in small animals for potential clinical application in 2002.
Subsequently, the safeties of inert agents, characteristics of distribution and
toxicity of potential therapeutic agents in the brainstem of small animals and
non-human primates have been studied. is approach has also been used
safely in a small number of patients with brainstem diseases. ese studies
showed that CED does not cause clinically relevant mechanical injury to the
brainstem and this approach has a promising therapeutic application in humans.
In clinical practice, image-guided frameless stereotaxy can be utilized to target
the brainstem in children for biopsy or cannula insertion with high accuracy
and low risks of temporary or permanent morbidity. ese will help establish
CED as an accepted drug delivery method in the treatment of DIPG.
erapeutic Ecacy of CED
CED of chemotherapeutic molecules has shown considerable promise in phase
I and phase II clinical trials in patients with recurrent malignant gliomas.
However, phase III results are less encouraging. CED in the treatment of DIPG
has produced encouraging results in preclinical studies. A few phase I trials of
CED in DIPG are recruiting patients or in the planning stage.
Several factors that are critical in achieving good therapeutic ecacy require
further elucidation. The convective force used in CED facilitates drug
distribution to larger volumes of brain tissue. However, malignant gliomas may
contain areas of brosis and necrosis, especially after receiving external beam
radiation therapy, which is currently part of the standard of care. CED, as an
investigational therapy, usually is not started until the completion of radiation
therapy. e brosis and necrosis may cause chaotic pressure gradients within
the tumor and therefore an unpredictable distribution of the drug. Even within
the peritumoral margins, targeting inltrating tumor cells may be limited by the
Chapter 17: Convection-Enhanced Delivery in DIPG
258
259
Chapter 17: Convection-Enhanced Delivery in DIPG
normal anisotropy of the brain tissue resulting in preferential ow of uid away
from the intended target. Furthermore, the presence of areas of disrupted BBB
either by the pathological changes or by previous treatment such as radiation
therapy may increase eux of drugs out of the CNS. A better understanding
of drug distribution will become a critical part of evaluating future studies
employing CED. Another concern is that the drugs infused in a single session
may maintain their therapeutic concentration for a period too short to be
eective before being cleared out of the target region. Once we have a better
understanding of drug distribution and clearance, other unsolved questions
including optimal catheter design and placement, infusion rate and duration,
and the benet of repeat infusions can be better addressed.
e use of targeted macromolecules allows for either intratumoral or peritumoral
treatment in malignant gliomas. Some of these agents may not be specic
enough, potentially leading to injury to normal tissue. is was seen with IL4-
PE, which initially started at a concentration of 2µg/ml. e potential benet
of targeting multiple molecules by combining dierent recombinant toxins, or
combining these agents with other chemotherapies, remains unknown. Despite
these limitations and uncertainties, signicant responses have been observed in
all of the CED clinical trials described below.
CED Clinical Trials for the Treatment of Brain Tumors
eoretically, any antineoplastic agent can be delivered through CED for the
treatment of brain tumors, including standard chemotherapeutic agents and
novel macromolecules such as monoclonal antibodies and viral vectors. One
unresolved issue is that CED, in its current form, is a surgery and typically
performed as a single session. It is unknown how long the infused drugs remain
at therapeutic concentrations after a single session of CED. Imaginatively, it
is more like a bolus dose, and predictably only a portion of the cancer cells
are killed by such a bolus dose and the remaining cancer cells will continue to
grow, ultimately resulting in failure of treatment.
For various reasons, most standard chemotherapeutic agents do not cross the
BBB in sucient amounts to have a signicant eect on the cancer. CED
of such small molecules showed that these agents have observable antitumor
responses. However, more neurological complications have been observed when
these agents were delivered via CED compared to systemic chemotherapy. ere
are eorts to improve formulations of these agents for local delivery to reduce
neurotoxicity and enhance therapeutic response. ese eorts, if successful, will
make CED of small chemotherapeutic molecules applicable on a larger scale.
More eort is focused on delivering recombinant toxins via CED in the
treatment of brain tumors. ese toxins are recombinant proteins and have
two components, a targeting moiety, typically a monoclonal antibody or a
ligand to an over-expressed cell membrane receptor, and a toxin, which can be
bacterial toxins. Bacterial toxins frequently utilized in recombinant toxins are
Pseudomonas exotoxin (PE) and Diphtheria toxin (DT). ese polypeptide toxins
have strong cytotoxicity against mammalian cells by inhibiting protein synthesis.
ey do not show selectivity in killing cancer cells over normal cells. But by
attaching them to a targeting moiety directed to cancer cells, the recombinant
toxins can become highly selective in killing cancer cells while sparing normal
cells. For this purpose, these bacterial toxins have been genetically modied
to make them easier to attach to targeting moieties. Genetic modication
also reduces the activity of these toxins to give a wider therapeutic window.
One targeting moiety widely studied for adult malignant brain tumors is
interleukin-13 (IL-13), because the IL-13 receptor is known to be over-expressed
in a high percentage of these tumors. Binding of a recombinant toxin on the cell
surface triggers internalization of the toxin, which enzymatically arrests protein
synthesis and ultimately causes cell death. Several recombinant toxins have been
utilized in clinical trials for adult malignant brain tumors delivered via CED.
ese toxins are attractive in that they have strong cell-killing capabilities and
resistance rarely develops.
Transferrin-CRM107
Several recombinant toxins have reached the stage of clinical study. e rst
cytotoxin that was used in brain cancer therapy via CED was Transferrin-
CRM107, a thioether conjugate of human transferrin and CRM107, a mutant
form of Diphtheria toxin. e compound was developed by a group led by
Richard Youle at the NINDS and is commercially available as TransMID™
from Celtic Pharma (HM, Bermuda). Transferrin-CRM107 targets tumor
cells by binding to the transferrin receptor, which is over-expressed on rapidly
dividing cells.
In a multicenter, open label phase II clinical trial, 44 adult patients received
intratumoral CED at 0.67µg/ml of Transferrin-CRM107 delivered directly into
the tumor bed. Numerous signicant clinical responses were observed. Of the
34 evaluable patients, ve had a complete response and seven a partial response.
e median survival for all 44 patients was 37 weeks. However, the tumor-
selectivity of this recombinant toxin is not high, shown by its toxicity to normal
tissues. In eight of the patients, increased cerebral edema was noticed. ose
with clinical neurotoxicity also had MRI changes suggestive of microvascular
Chapter 17: Convection-Enhanced Delivery in DIPG
260
261
Chapter 17: Convection-Enhanced Delivery in DIPG
injury, perhaps related to the higher levels of transferrin receptors on normal
blood vessel walls. A phase III multicenter, randomized study in recurrent,
nonresectable glioblastoma multiforme (GBM) was opened but withdrawn
prior to patient enrollment due to the toxicity data from the phase II trial.
IL4-PE
Another recombinant toxin clinically examined is IL4-PE, which is commercially
labeled as NBI-3001 (Neurocrine, San Diego, California) and PRX321 (Protox
erapeutics, Vancouver, British Columbia, Canada). More accurately called
IL-4(38–37)-PE38KDEL, the agent uses a mutant interleukin-4 (IL-4) as the
targeting moiety and a modied Pseudomonas exotoxin as the cytotoxic eector.
A phase I study of intratumoral CED of IL4-PE started at a concentration
of 2µg/ml and was dose escalated to determine the maximum tolerated dose
(MTD). Drug-related grade 3 or 4 CNS toxicity was seen in a total of 39% of
patients in all groups, and no systemic toxicity was seen. A phase II, multicenter
randomized study of intratumoral IL4-PE followed by tumor resection between
2 and 7 days after the completion of toxin infusion enrolled a total of 30 adult
patients. e accrual was completed in 2003 and the objective clinical responses
were not as good as Transferin-CRM107. A phase II trial of CED of IL4-PE
with real-time imaging for therapy of recurrent glioblastoma (the study is
referred to as CLARITY-1) has been approved but not recruiting patients as
of November 2008, the last time the status of the trial was reported. ere are
no plans for a phase III study.
TGF-α-PE38
TGF-α-PE38 is another recombinant toxin that entered clinical phase. It
is labeled as TP-38 commercially (TEVA Pharmaceuticals, North Wales,
Pennsylvania). TGF-α-PE38 is composed of transforming growth factor-α
(TGF-α), a native epidermal growth factor receptor (EGFR) ligand, and a
38kD fragment of the Pseudomonas exotoxin. TGF-α-PE38 binds to the EGFR,
which is over-expressed in the majority of GBM and is naturally present in
many normal organs.
Moderate or better responses were recorded in several patients in clinical trials.
A phase I study of intratumoral and peritumoral infusion of TGF-α-PE38 was
performed in 20 patients with recurrent malignant glioma with a concentration
escalation of 0.025 to 0.1µg/ml. Two catheters were initially placed during
tumor resection and then a total volume of 40 ml was infused. TGF-α-PE38
was well tolerated and a MTD was not established. At the completion of the
study, four patients had no recurrence of tumor over 55 weeks after treatment.
e overall median survival for all patients being treated was 28 weeks. For
those without radiographic evidence of residual disease at the time of therapy,
the median survival was 33 weeks. One GBM patient remains alive and without
progression more than 211 weeks after CED therapy, and another GBM patient
went 198 weeks without progressive disease after a nearly complete response
to TGF-α-PE38 and remains alive more than 260 weeks from CED therapy.
In the majority of patients imaged using SPECT, infusate distributions were
signicantly inuenced by leakage and failed to produce any signicant intra-
parenchymal distribution. is highlights the importance of accurate catheter
placement and drug distribution monitoring.
A phase II multicenter randomized study was conducted in adults with
recurrent GBM. Patients were randomized into two groups treated with
peritumoral CED of 0.05 or 0.1µg/ml of TGF-α-PE38. e total volume
infused was approximately 40 ml. Post-infusion MRI changes were seen 1 to
4 months after treatment, geographically associated with the site of catheter
placement. ese changes usually resolved by 20 weeks post-treatment. ere
were no grade 3 or 4 toxicities related to TGF-α-PE38. Only 20% of patients
retained the cytotoxin within the tumors by imaging. A phase I/II clinical trial
evaluating TGF-α-PE38 in treating young patients with recurrent or progressive
supratentorial high-grade glioma was terminated prematurely. Further clinical
trials are pending resolution of issues encountered in the phase I and II trials,
with catheter placement and infusate leakage as the most important concerns.
IL13-PE38
IL13-PE38 was developed by a research group led by Waldemar Debinski
in the mid-1990s. It is a recombinant toxin consisting of human IL-13 with
PE38QQR, a 38kD fragment of the Pseudomonas exotoxin. It is labeled
commercially as Cintredekin Besudotox by NeoPharm (Lake Blu, Illinois).
High levels of the IL-13 receptor have been found in more than 90% of
glioblastoma, whereas expression of the receptor in the normal brain is not
present or at low levels. is toxin demonstrated ecacy in several preclinical
GBM models before moving into clinical study.
Intratumoral and peritumoral CED of IL13-PE38 has been investigated in four
separate phase I studies. In the largest peritumoral phase I study, a maximum
tolerated concentration of 0.5µg/ml was observed. In this four-stage study,
histological ecacy, maximum tolerated concentration and maximum infusion
time were assessed. e nal stage explored the stereotactic placement of
catheters after tumor resection to improve targeting the peritumoral brain tissue.
Chapter 17: Convection-Enhanced Delivery in DIPG
262
263
Chapter 17: Convection-Enhanced Delivery in DIPG
A total of 51 patients with malignant gliomas were treated including 46 patients
with GBM. IL13-PE38 and procedure-related adverse events were primarily
limited to the CNS, including those associated with increased edema. With the
administration of steroids, all patients tolerated infusions of 40ml through 2
to 3 catheters lasting up to 6 days. e maximum tolerated concentration was
0.5µg/ml and tumor necrosis was observed at this concentration. ere were
no grade 3 or 4 adverse events associated with drug infusion at concentrations
lower than 0.5µg/ml, and no systemic toxicities were observed. Delayed
radiographic changes were observed in some patients 2 to 4 months after therapy,
which responded to steroids and may represent an inammatory response or
nonspecic activity.
e overall median survival for GBM patients was 42.7 weeks. Catheter
placement was variable in the early portion of the study, with some catheter
tips placed in CSF spaces. Catheter placement was correlated with survival. e
27 GBM patients with two or more catheters placed optimally without loss
of drug into the CSF compartment had a median survival of 55.6 weeks with
follow-up extending beyond 5 years, and 5 of these patients (18.5%) survived
beyond two years after a single treatment. ese trials showed that most of
the eective drug deliveries were achieved by infusing into the parenchyma
surrounding the gross total resection cavities rather than into the remaining
tumors themselves. ey also demonstrated that the chance of successful delivery
without reux or leakage was enhanced if the catheter tip was at least 2cm deep
from the last traverse pial surface and 5mm from the nearest non-traverse pial
or ependymal surface.
ese encouraging results led to a phase III multicenter, randomized study
(known as the PRECISE study) in patients with rst recurrent GBM. e
patients were randomized 2:1 to surgery followed by peritumoral infusion of
IL13-PE38 versus surgery and Gliadel wafer (MGI Pharma, Inc., Bloomington,
Minnesota) implant. Gliadel wafer contains carmustine (bis-chloroethyl-
nitrosourea [BCNU]) and is approved by the Food and Drug Administration
(FDA) as a standard therapy for GBM following surgical resection. Fifty-two
medical centers participated in this trial worldwide. Total enrollment was
targeted at 300 patients to demonstrate a 50% improvement in overall survival
in the experimental arm. Enrollment was completed in December 2005.
Analysis of follow-up data showed that this goal was not achieved. e median
survival of the 184 patients in the CED arm was 36.4 weeks compared to 35.3
weeks for the 92 patients in the control arm. When the dataset was restricted
to sites having enrolled more than six patients progressing to drug delivery,
the results are more encouraging. In this case, the CED arm had an overall
survival of 46.8 weeks versus 41.6 in the control arm, even though statistical
analysis showed that this cannot be said with sucient certainty. However, it
is signicant that progression-free survival was 17.7 versus 11.4 weeks in favor
of CED. e investigators believe poor drug distribution in some patients is a
major factor that adversely aected the therapeutic response. e trial implies
that a uniform method must be applied in participating centers to ensure exact
and reproducible drug delivery. Future trials will probably benet from improved
catheter placement, drug distribution and screening of expression level of IL-13
receptor chain α2 (IL-13Rα2). IL-13Rα2 is expressed specically by glioma
cells. e next generation toxin has been developed to bind the tumor-specic
IL-13Rα2 rather than the IL-13 physiological receptor, and should be studied
clinically.
131
I-chTNT-1/B mAb
131
I-chTNT-1/B mAb is an
131
I-labeled humanized murine monoclonal antibody
(mAb). It binds to a universal intracellular antigen, histone H1. Histone H1
is in the assembled DNA double strand and is exposed and accessible for
antibody binding in the necrotic core of solid tumors. is antigen provides
an abundant insoluble anchor for the mAb.
131
I emits γ rays with suciently
high energy to penetrate and kill adjacent tumor cells. From the principle of
how the drug was designed,
131
I-chTNT-1/B mAb is not as specic as those
targeting specic receptors (e.g., the EGFR or IL-13 receptors) expressed by
tumor cells, but rather delivers cytotoxic radiation to the tumor mass as well
as to tumor cells invading the surrounding tissue. “TNT” in the name of the
agent stands for “tumor necrosis therapy.”
131
I-chTNT-1/B mAb is commercially
labeled as Cotara (Peregrine Pharmaceuticals, Tustin, California). e eect of
131
I-chTNT-1/B mAb in patients with malignant gliomas was investigated in
several clinical studies. e results of two non-randomized, open-label studies
have been published: a phase I study in 12 patients with recurrent anaplastic
astrocytoma (AA) and GBM, and a phase II study in 39 patients with newly
diagnosed or recurrent malignant gliomas.
e 51 patients enrolled in the two studies included 37 recurrent GBM, eight
newly diagnosed GBM and six recurrent AA. All patients had previously
undergone radiation therapy, 42 had previously undergone at least one surgery
and 31 had a chemotherapy regimen. More than half of the patients (53%) had
a tumor volume of ≥ 30 cm
3
. One or two catheters with slit openings near the
closed distal end were placed with tips at or near the center of the enhancing
tumor.
131
I-chTNT-1/B mAb was infused using CED over 1 to 2 days at a
Chapter 17: Convection-Enhanced Delivery in DIPG
264
265
Chapter 17: Convection-Enhanced Delivery in DIPG
rate of 0.18ml/h. In the rst six patients, 1.5mCi/cm
3
clinical target volume
(CTV) was prescribed, which was calculated to deliver a dose of 137 Gy. For
subsequent patients, the dose was based on tumor size and the prescribed activity
was 0.5 – 3.0mCi/cm
3
administered in 1 or 2 infusions.
e phase I study showed that more than 130 Gy could be delivered to the
tumor with 34 ± 9% dose retention at 24 hours and a biological half-life of
46 ± 16 hours. Imaging and dosimetry studies on a subset of six malignant
glioma patients in the phase II study showed that infusion of 13.2 – 71.1mCi
of activity produced a calculated absorbed dose of 55 – 135Gy.
Treatment-emergent, drug-related central nervous system adverse events
included brain edema (16%), hemiparesis (14%) and headache (14%). Most
of these were reversed by corticosteroids. Systemic adverse events were mild.
Treatment with
131
I-chTNT-1/B mAb in the phase I study resulted in three
of nine GBM patients having stable disease at 60 days, and all nine patients
with progressive disease at 90 days. e median time to progression (MTTP)
and median survival time (MST) were 8.7 and 27.3 weeks, respectively. Of the
three patients with AA, one achieved a partial response and the other two had
stable disease 90 days after treatment. e 28 recurrent GBM patients in the
phase II study had an MTTP of 8.4 weeks (historical control 8.0 weeks) and
an MST of 23 weeks (historical control 24 weeks).
e phase II study contained patients with more diverse conditions. In an eort
to “normalize” ndings in this study, ecacy data from a subset of 12 recurrent
GBM patients who received a total activity between 1.25 and 2.5mCi/cm
3
,
which was considered a therapeutic window based on ecacy versus toxicity,
were examined. e median survival for these patients was 37.9 weeks. In
addition, seven of the 28 recurrent GBM patients and one of the three recurrent
AA patients survived for more than one year. Further research is required to
determine the value of
131
I-chTNT-1/B CED in these patients.
Two other phase I trials of
131
I-chTNT-1/B CED in patients with recurrent
or relapsed GBM have been completed recently and the results have not been
published. A dose conrmation and dosimetry phase II study for GBM patients
at rst relapse is ongoing. e dose is a single 25-hour infusion of 2.5mCi/cm
3
CTV. Brief interim results for a subset of 14 patients were reported in October
2010 and the median overall survival was 86 weeks.
Current and Upcoming CED Clinical Trials in DIPG
ere are no completed CED clinical trials for DIPG and only a small number
of CED trials for DIPG are under way or in the planning stage. is is in
contrast to the application of CED in the treatment of adult malignant gliomas,
where a number of clinical trials have been completed as summarized above.
Institutions sponsoring CED trials for DIPG have spent signicant eorts
in studying the safety of CED into the brainstem in small and large animals,
including non-human primates.
CED of IL13-PE38 for DIPG
e NINDS is sponsoring a phase I clinical trial led by Dr. Russell Lonser,
using CED to deliver IL13-PE38QQR into DIPG [Fig. 1a, 1b]. is study
started recruiting patients in 2009 and is expected to nish in early 2013. It
is an open label dose escalation safety study. IL-13 is an immune molecule
normally occurring in the body. About 90% of malignant gliomas have high
levels of IL-13 receptors while the normal brain tissue has only a low level of
these receptors. e experimental drug, IL13-PE38QQR, which combines the
modied PE with human IL-13, has been discussed above.
is study recruits patients 3 to 17 years of age with DIPG or supratentorial
high-grade glioma that have not responded well to standard radiation therapy.
20 patients are expected to enroll in this study. e planned doses are 0.125,
0.25 and 0.5µg/ml. Safety and tolerability are the primary endpoints with
secondary endpoints including imaging changes and treatment responses.
CED of
124
I-8H9 for DIPG
Memorial Sloan Kettering Cancer Center and Weill Cornell Medical College
are conducting a phase I clinical trial led by Dr. Mark Souweidane, using CED
to deliver
124
I-8H9 for the treatment of DIPG. is study is ongoing and
expected to be completed in 2014. is is an open label dose escalation safety
study.
124
I is a radionuclide with a half-life of 4.18 days. It emits γ rays and
positrons, both of which having energies high enough for therapeutic purposes.
Positrons will eventually be annihilated in the tissue resulting in the release of
photons that are detected in PET imaging. 8H9 is a monoclonal antibody that
binds to membrane protein B7-H3, which is expressed in high levels in most
DIPG but not by normal brain tissue. In principle, this antibody conjugated
to
124
I potentiates the antineoplastic eects of the radionuclide by directing
therapeutic irradiation preferentially to cancer cells.
is study recruits patients with DIPG ages 3 to 21 years old. e enrolled
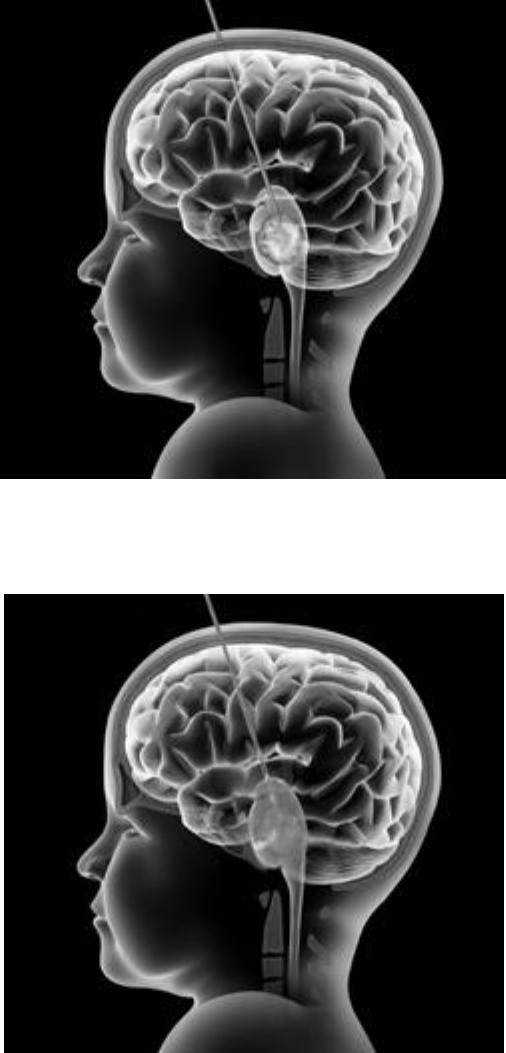
Chapter 17: Convection-Enhanced Delivery in DIPG
266
267
Chapter 17: Convection-Enhanced Delivery in DIPG
patients will have undergone standard external beam radiation therapy but have
not shown signs of progression. A maximum of 24 patients will be enrolled in
this phase I study. e planned doses are 0.25, 0.5, 0.75 and 1.0mCi of the
radio-antibody
124
I-8H9. Safety and tolerability are the primary endpoints.
Uniquely, this study uses PET to image drug distribution and calculate radiation
dose, which will provide invaluable information to correlate with tolerability
and therapeutic response. e usefulness of other imaging modalities in CED
planning in the brainstem will also be assessed as a secondary objective.
Future Directions
CED of therapeutic agents in the treatment of malignant brain tumors has shown
considerable promise in preclinical and some clinical studies. Future advances
will occur on two fronts: 1) the development of more eective therapeutic
agents for delivery via CED and 2) the improvement of the technique of CED.
A promising advance in the development of therapeutic agents for the
treatment of DIPG is the recent molecular characterization of this tumor.
ree groups independently discovered that the platelet-derived growth factor
receptors (PDGFR) are over-expressed in the majority of DIPG. erapeutic
agents targeting the PDGFR signal transduction pathways will be studied for
therapeutic ecacy. ese include anti-PDGFR antibodies and inhibitors of
the receptor tyrosine kinase (RTK) and downstream pathways. Another over-
expressed growth factor receptor is the EGFR. Like in the PDGFR pathway,
agents targeting the EGFR pathway include anti-EGFR antibodies and
inhibitors of the RTK and downstream pathways. e Sonic hedgehog (Shh)
pathway is over-activated in many cancers, including malignant gliomas. Its
study in DIPG is less in depth than in adult malignant gliomas. Inhibitors of
this pathway could also be potential therapeutic agents. Like in adult malignant
gliomas, IL-13Rα2 is highly expressed in DIPG therefore recombinant toxins
using IL-13 as a targeting moiety are also potentially eective therapeutic agents
for DIPG. e safety of CED of IL13-PE38QQR in the brainstem has been
investigated by the Souweidane group in preclinical studies and a clinical trial
sponsored by the NINDS is studying this agent in DIPG patients. Even though
biopsy of DIPG is far from being routine, when these targeted therapies based
on molecular proling of tumors come to clinical use, it would be ideal for the
tumor to be pre-screened for the targets that the drugs are designed for.
Increasing evidence shows that each individual tumor harbors multiple
mutations. For instance, there are on average 60 mutations per glioblastoma.
ere is no reason to believe DIPG contains a much smaller number of
Figure 1a:
Illustration demonstrating convection-enhanced delivery (CED) in the treat-
ment of diuse intrinsic pontine glioma (DIPG). An infusion cannula is inserted through
the transfrontal approach into the pons. e tip of the cannula will be at, or near the center
of the tumor. is is achieved by image-guided high-precision stereotaxy.
Figure 1b:
With the cannula in place, drugs are infused into the pons driven by a preci-
sion pump. Ideally, the drug infused area encompasses the tumor and the surrounding
inltrated area.
Chapter 17: Convection-Enhanced Delivery in DIPG
268
mutations than other malignant gliomas. Targeting one therapeutic target rarely
causes death to 100% of the cancer cells. RTK, downstream and parallel signal
transduction pathways may be regulated in complex compensatory fashions that
reduce the chance of cell death when the tumor is treated by aiming at only one
therapeutic target. erefore it is not surprising that drug resistance has been
inevitable in almost all single-drug targeted therapies. We believe it is worthwhile
to characterize defects in parallel and downstream signal transduction pathways
and devise multi-targeting therapeutic regimens based on such characterization.
On the technical front of the delivery method, there is a need for better designed
cannulas and more accurate stereotactic placement of cannulas into the tumor to
achieve optimal drug distribution. e use of computer algorithms may help in
planning the cannula placement and infusion parameters by taking into account
anatomical structures and structural changes induced by the disease and prior
treatment. Perhaps more important, imaging should accompany CED to ensure
eective drug distribution and concentration as well as to determine how long
the therapeutic agents are retained in the tumor and tumor-inltrated brain
tissue in individual patients. is requires the improvement of current imaging
techniques or the development of new imaging methods.
As discussed above, the current single session CED is more like delivering
a bolus dose. Clinically feasible methods to deliver multiple cycles of CED
or continuous CED lasting up to several weeks are desired. is will require
the development and engineering of catheters suitable for these purposes and
desirably also pumps that can be embedded and allow patients to remain
ambulatory.
CED-based therapies will continue to evolve, with a need for additional
preclinical and clinical research.

269
Chapter 18: Vaccine Treatment Strategies
Chapter 18
Vaccine Treatment
Strategies
Christopher Moertel, MD
Over the last 40 years, incredible advancements have taken place in the treatment
of childhood cancer. e overall cure rate for childhood acute lymphoblastic
leukemia is approaching 90 percent and some pediatric cancers have exceeded
that mark. New treatment strategies for medulloblastoma, a malignant brain
tumor of childhood, have increased the cure rate while managing to decrease the
dose of radiation necessary for cure. e history of cancer therapy has seen rst
advancements in surgical techniques, then radiotherapy, then chemotherapy.
Despite improvements in surgery and radiation delivery for some types of brain
tumors, the survival rates for one type of brain tumor known as glioblastoma
multiforme (GBM) has changed little over the years. The incorporation of
temozolomide in the treatment of adult GBM was heralded as a great treatment
advance, but it did little more than extend survival by about two and one half months.
Temozolomide has been used to treat children with the same tumor, but little to
no benet has been seen. Specically, children with DIPG (which looks like GBM
under the microscope) have seen no benet from the addition of temozolomide, let
alone any chemotherapy added to classical radiation therapy. Hence, the need to
keep looking for new answers to old questions. Is there any way we can nd a new
tool to treat brain tumors like GBM and its pediatric equivalent, DIPG?
Tumor Immunology to Treat Cancer
We are in a new era in which we are witnessing a great leap in the knowledge of
chemical pathways at work in cancer cells and
are now able to create designer drugs known
as targeted therapies to attack these pathways
and, hopefully disable the cancer cell. Another
novel area for cancer therapy has been the
focus on tumor immunology. e hope of
Dr. Moertel is a Professor of
Pediatrics and Clinical Director of
the Pediatric Brain Tumor Program
at the University of MN, and
Children’s Hospital in St. Paul, MN.
Chapter 18: Vaccine Treatment Strategies
270
271
Chapter 18: Vaccine Treatment Strategies
tumor immunology is built on the premise of taking advantage of the body’s own
weapons to attack foreign invaders and tumor cells. Some of the immunologic tools
in this arsenal are antibodies, immune stimulators and vaccines.
Vaccines have been used in medicine for decades to boost the body’s own immune
response to protect against invading foreign diseases, particularly infectious diseases.
In this case , the vaccine is given prior to the individual becoming severely ill with the
disease—hopefully with the result that the individual only exhibits a very mild form
of the disease or no symptoms at all. ere are currently two vaccines available which
prevent virus infections leading to the development of cancer. e rst is against
hepatitis B, a virus that can cause liver cancer. e second is against human papilloma
virus that is designed to stop specic strains of the virus leading to cervical cancer.
Tumor immunotherapy to treat childhood cancer is no longer just theory and research
taking place in the lab. A recent breakthrough in the treatment of neuroblastoma—
another dicult childhood cancer to cure, employs an antibody against the tumor
cells. One monoclonal antibody, called chimeric (Ch) 14.18 attacks a molecule
on the neuroblastoma cell surface called GD2. e body can then recognize this
antibody-tumor cell complex and attack it using its own immune system to destroy
the cancer. To enhance this immunological response from the child’s own body, the
children are also given ‘immune stimulators’ to excite their own immune system
eliminating even more antibody-tumor cells complexes. e immune stimulators
include interleukin 2 and granulocyte-macrophage colony stimulating factor (GM-
CSF). Some children can have signicant side eects, but the benet of cure far
outweighs the risks in the setting of this otherwise lethal disease. is approach has
helped saved many more children’s lives. Indeed, antibody therapy has become part
of the standard of care for treating advanced-stage neuroblastoma.
Tumor immunology to treat brain tumors
Researchers hypothesizing about the possible use of vaccines to treat brain cancer
were concerned about the established belief that the brain was “immunologically
privileged.” at is, the immune system could do little across the blood brain
barrier. However, our laboratories here at the University of Minnesota, in
addition to other labs across the country, have shown that immune system cells
and antibodies can go into the brain fairly easily. In fact, it has been shown that
malignant brain tumors can train cells called myeloid (bone marrow) derived
suppressor cells to “dumb down” the immune system. A lot has been learned
about the immune system’s relationship with the brain and the body’s ability
to mount an immune attack within brain tissue. is body of science has
encouraged many to pursue immunotherapy strategies against brain tumors.
Recently, there has been signicant energy put into vaccine therapies for brain
tumors, with GBM being the main target.
One group at Duke University has studied an antibody against a protein found in
excess on the cell surface of brain tumors called Epidermal Growth Factor Receptor
(EGFR). Indeed, about 20% of GBMs have a mutant variant of EGFR called
EGFRvIII. So if EGFR is found in abundance on tumor cells and EGFRvIII is a
unique mutation on tumor cells, why not use this as a target for a programmed
immune attack? at is what the Duke group did. First, they conducted a clinical
trial combining their EGFRvIII protein with cells from the patients’ immune
system called dendritic cells (DC) and re-injected them into the patients with newly
diagnosed GBM. is led to an overall survival rate that was better than expected.
ey then went on to do a study in partnership with MD Anderson that used their
EGFRvIII vaccine combined with KLH, a protein that excites the immune system.
None of the patients’ dendritic cells were removed and re-injected in this study and
everyone received “standard” temozolomide in addition. e results of this trial were
somewhat encouraging. Immunotherapy for brain tumors burst onto the scene and
a great deal of enthusiasm and interest was created in the brain tumor community.
Currently there is one trial open at Stanford for newly diagnosed children with
DIPG that uses an EGFRvIII vaccine. is vaccine trial was initiated after Dr. Li
and colleagues reported 50% of DIPG tumors had EGFRvIII on their cell surface.
While the Duke group was working on their specic protein-based approach,
another group in California was using a patient’s own tumor cells to make a
vaccine. Dr. Linda Liau and colleagues resected the patient’s own tumor, cultured
it in the lab and separated proteins from the surface of the tumor cells. ey
then removed cells called mononuclear cells from the patient’s circulating blood
through a process known as apheresis and separated cells called dendritic cells
(DC). e patient’s own tumor surface proteins were then combined with his
or her DCs, incubated for up to an hour, and re-injected into his or her body.
is treatment is best called an acid-eluted glioblastoma multiforme peptide-
pulsed dendritic cell vaccine. Dr. Liau’s group showed that the vaccine was safe
and, resulted in longer survival for a number of patients. is work has now
been commercialized (Northwest Biotherapeutics, Inc.) and is available to adult
patients with GBM through a phase II trial at participating centers. is therapy
is not yet available for DIPG patients.
Another group led by Dr. Hideo Okada at the University of Pittsburgh has had
signicant experience with glioma-associated antigen-derived synthetic peptides
that excite cytotoxic T cells. Okada’s work also employs dendritic cells incubated
with the peptide and given along with a substance that excites the immune
system called Poly-ICLC (polyinosinic-polycytidylic acid). is peptide strategy
Chapter 18: Vaccine Treatment Strategies
272
is available only to individuals with a specic HLA subtype, A2. Unfortunately,
HLA-A2 is present in only 45% of the population. One of Dr. Okada’s vaccine
studies conducted with Dr. Regina Jakacki, who has participated in and run a
number of DIPG clinical trials, target DIPG specically. Dr. Okada has reported
(unpublished data) that at least one vaccine patient exhibited signicant tumor
regression after initial worsening of the MRI abnormality in the brainstem.
At the University of Minnesota, we have developed a dendritic cell vaccine
based on a brain tumor initiating cell (BTIC) line called GBM6. is cell line
has surface characteristics on the cells that match the surface characteristics of
initiating cells in many types of brain tumors, including glioblastoma. e cell
line also more closely mimics the growing conditions of human brain tumors
by being grown in 5% oxygen rather than the usual room air or 20% oxygen
seen in most lab environments. While our research is still early, we are in the
midst of a phase I trial that is showing that patients with brain tumors have
immune responses to our vaccine and may have tumor regression or stabilization
without any unusual toxicity. Indeed, the stronger immune responses have been
noted in our younger patients. Based on this early data, we are going on to start
a phase I trial targeting patients with newly diagnosed DIPG using a lysate of
our GBM6 cell line and topical Imiquimod, another immune system-enhancing
agent. Initially, we will be treating adult patients with GBM so that any unusual
toxicity will be dealt with before kids are exposed to this new treatment modality.
We feel this treatment approach has three specic advantages for the DIPG
population. First, there is no need to obtain tumor tissue to create the vaccine
(a nearly impossible task in patients with DIPG). Second, the vaccine is not
HLA restricted, as is the case in most peptide-based vaccines, being available to
the entire population. ird, this new vaccine trial will not use dendritic cells.
Hence, there will be no need for apheresis and all of the diculty that entails.
e common experience among those conducting brain tumor immunotherapy
trials is that minimal residual disease is optimal at the beginning of treatment.
Because DIPG cannot be surgically removed, we will need to depend on
radiation therapy to create minimal disease. Unfortunately, this means that
immunotherapy strategies will generally not be optimal for those whose disease
has relapsed, unless caught at a very early stage.
All of this is very early and may certainly end up not providing what we and all
parents of children with DIPG ultimately desire, but the early experience with
GBM immunotherapy tells us that there is a glimmer of hope for patients with
DIPG that has not been realized with classical radiation and chemotherapy–based
approaches. At the very least, we hope that new pathways to a cure will be revealed.

273
Chapter 19: DIPG and Tissue Donations
Chapter 19
DIPG and Tissue
Donation
Cynthia Hawkins, MD, PhD
Eric Bouffet, MD
Ute Bartels, MD
Before magnetic resonance imaging (MRI) became available, surgery was usually
recommended to conrm the diagnosis of brainstem tumors including diuse
intrinsic pontine glioma (DIPG) to provide both histological and prognostic
information. However, surgery was associated with signicant morbidity and the
benet of this approach was questioned when it became evident that the MRI
scan was able to provide images that were basically diagnostic of DIPG. erefore,
since the early 1990s, the frequency of biopsy has signicantly decreased and most
DIPG children are currently treated without histological conrmation.
Although this approach is the result of a consensus, over the years there has also
been increasing awareness that progress in the management of this deadly disease
will only occur with more biological information on DIPG. In this context, several
teams have explored ways of collecting tissue material, in particular at the time
of death, from limited brain or brainstem autopsies.
Early Experiences
Autopsy
Autopsy was a standard practice for in-hospital
deaths in many institutions until the late 20th
century. Autopsies were a standard procedure
in former times. They were performed
to identify the cause of death, and the
contribution of autopsies to the understanding
of many conditions has been significant.
However, there has been a steep decline in
Dr. Hawkins is a Principal
Investigator at the Arthur and
Sonia Labatt Brain Tumour
Research Center, an Assistant
Professor at the University of
Toronto, and Neuropathologist
at the Hospital for Sick
Children, Toronto, Canada.

Chapter 19: DIPG and Tissue Donations
274
275
Chapter 19: DIPG and Tissue Donations
autopsy rates over the last decades as, in many cases, the cause of the patient’s disease
was felt to have already been identied. A drawback to this practice, however, is that
it limits researchers' ability to study diseases at the tissue level particularly for diseases
such as DIPG, where no surgical procedures are ever undertaken. us, until recently
there has been no experience with systematic collection of brain or brainstem tissue
to advance research in DIPG. Now groups from Canada and the U.S. have reported
this experience and demonstrated the possibility to use postmortem material for
genome-wide studies and even for generating cell lines.
In the Toronto experience, the concept of autopsy was discussed by the treating neuro-
oncologist after radiation therapy, at the time of progression or later during palliative
care. Sometimes this discussion was initiated during a home visit. Explanation
included the dierent types of autopsies, in particular the possibility to restrict the
autopsy to the whole brain or to the brainstem tumor according to the family’s
preference. In cases where only the tumor was removed, a small biopsy of the frontal
normal brain was also performed. To obtain appropriate tissue, in particular RNA
(ribonucleic acid), the autopsy needs to be done relatively early following death. In
the St. Jude experience, there was minimal RNA degradation when the autopsy was
performed within 5 hours of death; however, the quality of RNA dropped signicantly
beyond that delay. Similarly the possibility to grow cell lines from tumor tissue is
strongly related to minimizing the delay between the time of death and autopsy.
Interestingly, in both the Canadian and U.S. experiences, most patients died at
home (88% and 84%, respectively). If a family or patient consented to an autopsy,
the process was carefully organized ahead of time in order to avoid any delay in the
funeral. At the time of death, a transportation service was arranged to pick up the
body and transfer it to the academic center or to a local hospital where the autopsy
was performed within hours. e autopsy itself was carefully performed in order to
avoid any visible scars, in particular in the context of viewing visitations and open
casket services, which are an important part of bereavement in the North American
culture.e body of the child was brought back to the funeral home. is organization
allowed privacy for the family and a timely funeral without delay.
e issue of the consent for post mortem tissue collection has been described in
detail in one publication from the Toronto
group. In this experience, 10 out of 21 parents
who were approached gave their approval. For
the 10 families who consented, their main
motivation was to support research and to
contribute to breakthroughs in scientific
research on DIPG to benet future children.
In this experience, 2 children (11 and 10 years old) expressed their own wish to
donate their brain for research purposes. e main reasons for declining autopsies
were ethical and/or religious reasons or related to the level of emotional distress. Only
one family felt upset by the request for brain donation. In all cases, when an autopsy
was performed, a face- to-face meeting took place between the treating physician and
the parents once the results of the autopsy were available. No parents expressed any
regret from having given consent. All expressed their hope that their child’s death
may help research and contribute to the development of new treatments.
e scientic results of the research conducted on postmortem material have been
reported since 2009 in several important publications. Researchers have been able to
identify a number of potential targets for new treatments. is work has also shown
that DIPG is not a single entity and that dierent types of treatments may be needed
according to the underlying biology of the tumor.
Stereotactic biopsies
In parallel, other teams have decided to reintroduce the concept of biopsying DIPG
at the time of presentation. e neurosurgical team from Necker Hospital in Paris
has reported on the feasibility and safety of stereotactic biopsies of patients with
newly diagnosed DIPG. In their initial report, 2 out of 24 patients presented with
a transient decit associated with the procedure. ey have subsequently reported
and updated their experience and have conrmed the safety and feasibility of DIPG
biopsy in more than 80 patients. is work has also provided new information
regarding the biology of DIPG. Interestingly, the results of studies conducted with
tissue obtained at the time of diagnosis or at autopsy (therefore after radiotherapy
in most cases) do not dier considerably.
What Have We Learned So Far About DIPG From Autopsy
Studies?
ree genomic studies have now been published on autopsy series of DIPG.
While still somewhat limited when compared to genomics studies conducted
in adult cancer, several important conclusions can be drawn from the data
available from these DIPG studies which can help in the development of future
clinical trials. First, the studies support dierences at both the copy number and
expression level that distinguish pediatric
DIPG from both adult and pediatric
glioblastoma multiforme (GBM) elsewhere
in the brain. is conrms that DIPG must
be considered as a separate biologic entity
Dr. Bouffet is the Director of the
Neuro-oncology Program and
a Senior Associate Scientist in
the Research Institute at the
Hospital for Sick Children, as well
as Professor of Pediatrics at the
University of Toronto, Canada.
Dr. Bartels is an Oncologist in
the Neuro-oncology Program at
the Hospital for Sick Children,
and Assistant Professor at the
University of Toronto, Canada.
Chapter 19: DIPG and Tissue Donations
276
277
Chapter 19: DIPG and Tissue Donations
for the purpose of clinical trial design.
Second, receptor tyrosine kinases (RTKs) appear to be upregulated at the
genomic or expression level (or both) in the majority of pediatric DIPGs. e
most commonly amplied RTK in pediatric DIPG is PDGFRA, occurring at
the genomic level in at least 30% of DIPGs with an even larger number showing
over-expression at the RNA and protein levels. Gain of EGFR does not appear to
be a frequent event in pediatric DIPG. However, two other RTKs are reported
to be frequently gained in DIPGs: MET and IGF1R. Interestingly, in many
cases more than one RTK is amplied in the same tumor, a nding that may
have implications when using single RTK-inhibitors.
Based on their work on stereotactic biospies, the team for Necker (Paris)
has described two distinct subgroups of DIPG. e rst subgroup shows
mesenchymal and pro-angiogenic characteristics. e second subgroup displays
oligodendroglial features, and appears to be driven by PDGFRA. is later
group had a signicantly worse outcome and shorter survival expectancy. is
suggests that dierent treatment strategies may be needed as DIPGs do not
appear as a uniform disease.
Current Situation
ese experiences have generated signicant hope and enthusiasm in the DIPG
community and also amongst parents and support groups. In April 2009, the
FDA held an open public hearing specically to discuss the science and ethics
regarding pediatric DIPG biopsy in the United States. e purpose of the FDA
hearing was to address a research initiative proposed by a U.S. based children’s
hospital that wanted to biopsy newly diagnosed DIPG children to compare
similarities between their tumors and pediatric cerebral glioblastomas. Several
issues were addressed during the 2009 FDA hearing including the concern of a
number of the panel members that the risk of biopsy of DIPG was not balanced
by certain benet for the individual child. As a way to bridge this issue, several
experts suggested that an initial eort be made to evaluate port-mortem tissue.
is would alleviate concerns regarding safety of biopsy and also make more
tissue available for research purposes. Several groups (Dana Farber of Boston,
UCSF of San Francisco, the National Cancer Institute of Bethesda, the Institut
Gustave Roussy of Paris) are currently working on biopsy driven protocols. In
these protocols, the results of the biopsy will be taken into account to stratify
patients and allocate their treatment accordingly.
Concurrently, an increasing number of institutions, including Sick Children’s
Hospital in Toronto, St. Jude Children's Research Hospital, and the National
Institutes of Health (NIH), already have research procedures in place for autopsy
tissue donations from families whose children were treated under their care or
in outside institutions.
Where Do We Go From Here?
Autopsy studies have triggered a tremendous enthusiasm in the pediatric neuro-
oncology community. Several protocols have been developed or are about to
open following the publication of the rst genomic studies that have identied
potential targets. However, most protocols target one mechanism only and it
is likely that the successful management of DIPG will require combination
therapies. In this context, recent development of genetically and histologically
accurate preclinical (animal) models is critical, as this will allow the testing of
targeted therapies that may eventually be brought to the clinic. A number of
models are currently being tested that may contribute to selecting the most
appropriate agents or combinations for future clinical trials. ere is no doubt
that the coming years will see an explosion of new DIPG protocols based on
the data generated by the collection of autopsy material. Everyone hopes that,
as a result, we will observe for the rst time a dierence in the dismal outcome
of children with DIPG.
Chapter 19: DIPG and Tissue Donations
278
279
Chapter 19: DIPG and Tissue Donations
Parent Perspectives
I know this is such a difficult thing to consider while a child is fighting, but
planning for tissue donation is not giving up in the fight. It is just one of the
many ways we can prepare for our children’s memories to endure and for their
lives to make a difference, even as we hope and plan to never reach the day.
None of Caleb’s doctors ever approached my wife or me about a donation
of Caleb’s tumor. Through reading the web blog of another family whose
son had passed away, I learned that they had donated his tumor and I
became aware of the medical value of the tumor. I would not have known
the tumor had any value, or that a doctor would want it, if I’d not stumbled
across that web page.
While we were on a routine monthly trip to the NIH, I raised the subject with
Caleb’s doctor. Caleb was still doing well, but I knew the prognosis of his
disease and I wanted to begin the discussion with his doctor. She seemed
hesitant to discuss it, but simply said that when the time was right, there were
some studies that could use the tumor and we could certainly discuss it later.
I also asked Caleb’s doctor in Houston about donating the tumor, wanting
to make sure they knew of my interest. Since my wife had not yet accepted
the possibility that Caleb could die, I did not discuss my questions with her.
When Caleb began to worsen, and it finally became clear that he would not
survive, I raised the topic with my wife. She was open to making the donation,
but wanted Caleb’s doctor to be at the hospital to receive him, and so I more
seriously pursued the topic with Caleb’s doctors at the NIH and in Houston.
They informed me that there were studies that could use his tumor, and they
would be glad to receive it. I know recent cases where parents selected the
studies and beneficiary institutions for the tumor tissue, but at the time, I
didn’t know this was possible. So we simply donated it to the hospital in
Houston where Caleb was treated, making sure our doctor at the NIH knew
of the donation and could obtain the tissue she needed for her studies as well.
I also talked with our funeral director early on. I was concerned about
Caleb having an open casket funeral, and I didn’t want to do something
that would disfigure him noticeably. Both the doctors as well as the funeral
home assured me this should not be a problem.
We signed the paperwork for the study, and there were no expenses at all
which were charged to us for either the surgery or the transportation of
Caleb’s body to the hospital for the removal of the tumor.
Caleb passed away at home, and we kept him there for a few hours afterwards.
Then, the funeral home brought him to the hospital for the removal of the
tumor. Caleb’s doctor agreed to my wife’s request and was present during
the tumor removal, to ensure he was lovingly taken care of. We did not see
Caleb again until he was embalmed and ready for viewing.He looked amazing,
and angelic. The doctors and the funeral director were right—there was
no evidence whatsoever of the tumor removal; they must have done a very
discreet incision on the back of his head (which was resting on the pillow).
He looked beautiful.
In hindsight, we would do this all over again. Knowing that Caleb might
help other children fighting the disease, even in his death, brought us some
peace. And, having the tumor removed from him was also important to us.
We wanted it gone, even if that were only possible in his death.
Five years have passed now, and there are many more options available.
Some parents have selflessly made themselves available to help others
through this experience, and I wish we would have had that help with our
decision. There are many studies, well known, which will accept tumor tissue
donations. Parents can have a say which studies benefit from the donation,
and the tumor can be divided among several studies. Every parent must
make their own decision, but for us, this was the best decision.
Probably mid-way through Mara’s diagnosis with DIPG we heard about
the need for tissue samples. For us, the decision to donate seemed easy and
quite frankly it seemed like the only way we could make a difference for
the future of this disease. To us, donating meant that if Mara were to pass
away, it would not be in vain, for perhaps someone else could live even
though she could not. Initially though, we prayed to be THE MIRACLE
and didn't want to think about crossing that bridge.
It wasn't until Mara's pediatric oncologist set up a palliative care meeting
for Mara at the end of August (approximately 1 month before her passing)
that the issue was discussed directly with them. As I recall, we were the
Chapter 19: DIPG and Tissue Donations
280
281
Chapter 19: DIPG and Tissue Donations
ones in this meeting to bring up tissue donation. As I look back, I was a
little surprised that our medical team never mentioned it to us directly.
However, I see their wisdom in it now. What kind, compassionate doctor
can tactfully ask a parent for a tumor donation when their child is still
living and the family is still fighting? So, my husband and I brought up
the issue of having Mara's tumor donated upon her passing as well as any
other organs that could be donated.
Mara’s doctor was honored that we would suggest donation. He explained to
us that we'd have to sign a consent form and we did that at Mara's next CT
scan, which was a few weeks later. The consent form we signed was a rather
generic form. It was a form that anyone would sign to release tissue, whether
it is due to the child's passing or just a simple donation derived from a biopsy.
We made it clear at this time that we'd like samples to be sent to St. Jude and
to the NIH, as well as the researchers at Seattle Children’s Hospital. He did
communicate with us the need to have Mara transported post mortem and
taken from our home up to Seattle (about an hour away). He told us that any
cost associated with the transportation would be paid for by the hospital.
As the week continued, we realized that we should probably at least touch
base with a funeral home to let them know about our situation and that
transportation would be an issue. Bryan made this excruciating call, an act I
can never imagine doing myself. It seemed against every parental inclination
to arrange for our child's death when she had not yet passed. This phone call
occurred only 2 days before Mara left her earthly home. The funeral home
assured us that this would not be a problem. We gave hospice the funeral
home information, which worked out well because when Mara did pass away
they took care of everything. We didn't have to think about it anymore after
this phone call. It made the decision much easier in the long run.
The day Mara passed, we received a phone call from her doctor and from
our nurse practitioner telling us that the autopsy was being performed and
that the transport went smoothly. They cried with us, and thanked us. Later,
we also received a note in the mail from Mara’s doctor thanking us once
again and letting us know that in his opinion, we did everything possible to
help Mara. Reassurances like that mean the world to grief-stricken parents.
In the end, nothing about this disease is easy, nothing. I understand that
the decision to donate is most personal in nature. However, the pride in
my heart for my daughter’s offering is beyond description. For us, this was
the best decision and will allow Mara’s legacy to live on.
We had already made arrangements to donate Caleb’s tumor for research.
So, after my husband and our other boys and our friends had had some
time with Caleb's body, my husband called our oncologist to let her know of
his death and to make plans to meet her at the hospital. Our funeral home
director is our neighbor and was prepared to hear from us. He came to the
house when my husband was ready. They rode to the hospital with Caleb's
body and delivered it to our oncologist (with our friends following behind).
After my husband was sure that our oncologist had Caleb's body and was
prepared to supervise the harvesting and preservation of the tumor tissue,
he left the hospital with our friends. After the autopsy was complete, the
funeral director took Caleb's body back to the funeral home.
Our son Andrew died on December 4, 2009. We donated his tumor to his
neuro-oncologist at the NIH, and tissue was shared with Hopkins and
Children's National Medical Center.
I have been helping with DIPG and other brain tumor tissue donations
since November, 2008. As a result of tumor tissue donation over the past
few years, researchers are beginning to have an understanding of DIPG
biology which means that treatments can now actually be developed and
chosen in a scientific way. Though we do not yet have a cure for DIPG, we
are headed in the right direction. We could not say this in October 2007
when my son was diagnosed.
It was almost seven months after Ethan passed when I happened to email
his oncologist. His oncologist shared information with us that he had
received from a researcher at Texas Children’s. I wanted more information,
so I emailed the researcher directly. I wasn't sure I would hear back, but he
was very willing to share with me the progress of his research with Ethan's
tumor and the mouse model he had created with it.
I believe in miracles but I am also a realist. As we sat in the exam room
listening to the doctor tell us our daughter had a brain tumor for which
there was no real treatment much less a cure, I remember thinking I want
this thing out of her head. If we don’t get our miracle someone has to
Chapter 19: DIPG and Tissue Donations
282
283
Chapter 19: DIPG and Tissue Donations
study this beast so no one else has to do this. I had no illusions that our
donation in and of itself would be the magic bullet for DIPG patients but
Hope was special and her life mattered. Her grey matter would be studied
and knowledge could be gained.
While I am positive that our doctor would have brought tumor donation to
our attention at the time he thought appropriate, I beat him to it. I always put
reality in the driver’s seat with hope firmly strapped into the passenger seat.
I wanted to know up front what all my options were. When he said, “Don’t go
online I will answer your every question,” I raced to the computer. I trusted
him completely but I needed to know ahead of time what potentially could
or would happen. We had conversations throughout Hope’s battle regarding
tumor donation. These conversations were always welcomed by our team
but were initiated by me. I felt compelled to be sure they understood how
important it was to me. They felt bound by compassion to be gentle with me.
As Hope appeared to be nearing the end of her life we had our most serious
conversation regarding our donation. Hope’s doctor assured us that he
would stay with Hope during the entire autopsy and made sure her “blankie”
was nearby. He asked us for permission to share her tumor with other
researchers, which we happily agreed to. We did not ask where he would
send the tumor or how it would be used as we had complete confidence
in his professional judgment. We have no regrets. Hope’s tumor is being
studied at some of the finest institutions in the country.
If I had any suggestions to give, I would suggest that doctors be upfront
with families about tumor donations and for families to understand why they
need to do so. These relationships are built out of respect and honesty. It
is unfortunate that the time line for our kids is so short that doctors really
can’t afford to wait for the “right time.” Just like a clinical trial is fully
explained as an option, so should tumor donation be explained as an end of
life option. I wish I had known more about the need for live cell lines and
what they have to offer research. While I know that Hope’s tumor, having
been frozen, is offering much to the research community, it would have
made my heart happy to know that somewhere the evil thing that took my
child’s life was alive and potentially being used for the good of our kids.
Throughout my daughter’s battle with DIPG, I constantly struggled to
balance my ultimate hope that she would somehow survive, with my
acknowledgement of the reality of the disease and the likelihood that
she would not. By nature, I am a planner. Subconsciously, I think that I
somehow believe that if I can think through all of the possible outcomes
and scenarios and plan for all contingencies, then it will be easier to deal
with whatever actually happens when the time comes. Of course, I knew
that I could never prepare for my daughter’s death.
So, it wasn’t long after my daughter’s diagnosis that I learned about
tumor tissue donation and began to think about whether it was something
I would want to do. Our daughter’s medical team did not mention it for
a long time; in fact, they didn’t mention it until very close to the end. But
long before they brought it up, we had considered it. We learned of other
families who had donated, and how comforted they were in knowing that
perhaps their child’s death was not in vain—that the donation might help
another child someday. Throughout my daughter’s battle, my husband and
I occasionally discussed the issue. He was hesitant to do it, while I thought
it would be comforting. In the end, we discussed it with our clergy, other
parents who had donated, and with a few very close friends and family. We
were reassured that it would not affect our funeral plans or how long we
would be able to stay with our daughter once she had died. We also were
told that the logistics would be handled and that it would not require any
additional burden on our part at such a difficult time.
Despite all of that, ultimately we decided that donating our daughter’s tumor
tissue was not the right decision for us or for our daughter. It was not an
easy decision by any means; but it is one with which we are comfortable and
do not regret. Throughout the course of her battle, she had participated in
four clinical trials. She had sacrificed so much already in her short life. She
went through extra weekly pokes, prods, and tests, and never complained.
She had scars from various surgeries. She took drugs that caused her side
effects but gave her no benefit. She had suffered enough. My husband and I
also struggled with the thought of picturing our daughter after the donation.
We wanted to remember her as the beautiful little girl that she was, and
not be clouded with images of her “less than whole.” While I, no doubt,
would have found great comfort in possibly helping to find a cure through
donation, that comfort may not have outweighed the emotional distress that
such a donation may have caused me and my husband. Whatever parents
decide; it will be the right answer for them and their family. But parents
should know that it is okay if they choose not to donate.
Chapter 19: DIPG and Tissue Donations
284
Our decision to donate our daughter’s tumor tissue was easy in comparison
to the other decisions we had to make at that time. We had just been dealt
the news that her tumor had progressed and we were dealing with being
removed from a clinical trial, setting up hospice care, and looking for other
treatment options. Other parents of DIPG children mentioned they would
be willing to help set up the tumor donation. I asked my husband what he
thought and he said we should go ahead with it. I agreed. I contacted the
parent/advocate who had offered to help and we began the process. Of
all the decisions we had to make at that time, we gave that one the least
amount of thought. We did not realize the positive impact it would have.
Because our daughter’s condition deteriorated so quickly, our involvement
in setting up the donation was minimal. We made important choices, such
as where the tumor would ultimately be donated, but the DIPG mom who
was helping us let us know that she could take care of the logistics if we
did not have the time. We were able to choose our level of involvement so
that we could focus on our daughter during her last days on earth.
After she passed away, we signed some paperwork and the donation was
made. The donation did not disrupt our plans for her Catholic mass and
burial. We laid our daughter to rest tumor free. We opted to have her tumor
go to Dr. Monje at Stanford. Once the donation was done, I thought that
would be it. But Dr. Monje has graciously shared her progress in research
using Bizzie’s donation. She took the time to provide us with details about
our daughter’s tumor and how she was using the tissue. Within weeks of
our donation, Dr. Monje shared with us that Bizzie’s tumor was used to
confirm an important finding in DIPG research. To know that our decision
to donate has, along with other donations, facilitated a finding that could
help develop an effective treatment for future DIPG kids is something that
honors our little girl’s memory in a way that nothing else could. No amount
of fundraising could provide the same value. While we could not help our
daughter beat cancer, we can help other kids have a chance at growing up.
Brendan knew there was no cure and that he would die from the tumor. He
also knew that donating the tumor could lead to a cure in the future. His
tumor tissue was donated to Children's Hospital and a pathology report
was shared with us. His post mortem tissue is going to be useful for future
research.

285
Chapter 20: Organ and Tissue Donation
Chapter 20
Organ and Tissue
Donation by Pediatric
Brain Tumor Patients
Angela Punnett MD, FRCPC
Some patients with brain tumors and their families may wish to explore the
potential for organ and tissue donation following an expected or unexpected death.
is is a complicated decision and it is helpful when planning to understand the
background of organ donation in the context of a cancer diagnosis; issues specic
to patients with brain tumors; the process for donation; and implications of organ
donation at the time of death.
History of Organ Donation and Cancer
ere are a signicant number of patients awaiting solid organ transplantation at
any given time and 5% to 7% of these patients will die while awaiting transplant.
Lack of consent to organ donation by the population at large is the main limiting
factor to transplantation.
It is generally accepted that individuals with a diagnosis of cancer are not eligible to
donate their organs because of the risk of transmitting cancer to the organ recipient.
is risk of transmission has been documented since the early days of solid organ
transplantation, with reports of close to half of recipients developing the donor’s
cancer, resulting in death.
However, not all cancers are equally likely to
spread, and recognizing the need to balance
pre-transplantation life-threatening conditions
with post-transplantation malignancy risk,
an International Consensus document was
written in 1997 (Council of Europe, 1997).
Dr. Punnett is an Assistant
Professor in the Department of
Pediatrics at the University of
Toronto, and Program Director
of the Pediatric Hematology
Oncology subspecialty training
program at The Hospital for Sick
Children, Toronto, Canada.
Chapter 20: Organ and Tissue Donation
286
287
Chapter 20: Organ and Tissue Donation
e group of experts who crafted this document recommended consideration of
donors with primary brain tumors that very rarely spread outside of the central
nervous system (CNS).
Risk of Spread of Primary Brain Tumors
Spread of a primary brain tumor outside of the CNS, or extracranial
metastasis, has been considered unlikely because of anatomical and biological
features of the CNS itself. e CNS is a relative sanctuary with the so-call
“blood brain barrier” and has few lymphatic channels. As a brain tumor grows
it invades surrounding tissue, collapses existing blood vessels, and really has
nowhere else to go. However, studies have shown that brain tumor cells can
grow in other types of tissue and invasion of blood vessels and lymphatic
drainage has been documented.
e most important and consistent risk factor for extracranial spread is the
cell type and grade of malignancy. e tumors most likely to demonstrate
extracranial spread include ependymoma at about 6% of patients,
medulloblastoma at about 5% of patients, and glioblastoma at about 0.5%
of patients. ese numbers are based on relatively limited and older data
however, and are dicult to interpret. In addition, a number of other risk
factors for extracranial spread have been proposed, including duration of
disease, receipt of chemotherapy and/or radiation therapy, and history of
craniotomy and/or ventriculosystemic shunt.
Donor transmission of primary brain tumors
ere are a number of case reports in the literature documenting transmission
of tumors from donors with primary brain tumors to their organ recipients,
in some cases causing death. Recipients of heart, lung, or liver transplants,
were more likely to die from transmitted malignancy, as kidney transplant
recipients could be saved with chemotherapy or removal of the transplanted
kidney.
A number of transplant centers have reviewed their local experience in an
attempt to understand the risk of transmission. e resulting analysis showed
that donors with a history of a primary brain tumor accounted for 1% to
4% of all donors. Among those donors with a history of a primary brain
tumor, the risk of transmitting a tumor to an organ recipient was 0% to 3%.
Larger experience is available through mandatory reporting to national organ
transplant registries. In the United States, the United Network for Organ
Sharing (UNOS) has published its experience, which shows donors with
primary brain tumors consistently represent approximately 1% of all donors.
Out of 642 transplant recipients, 3 developed a fatal tumor from one donor
with glioblastoma multiforme (0.5% transmission rate). When comparing
survival curves for recipients of kidney and liver transplants from donors
with and without primary brain tumors, there is no dierence. Donors with
primary brain tumors represent 2.6%, 2%, and 1.5% of all donors in the
Australia/New Zealand, Czech Republic, and United Kingdom registries
respectively, with no cases of donor-derived malignancy reported.
Somewhat dierent data is available through the Israel Penn Tumor Registry,
an international, voluntary reporting registry based in the United States that
started in the very early days of transplantation. It is not possible to estimate
the incidence of transmission with this registry data, but it is possible to
look at risk factors for transmission. Among 62 organ recipients from 36
donors with primary brain tumors, 14 (23%) developed a tumor; almost
half of the donors had glioma/glioblastoma. Risk factors for transmission
from this data include the presence of ventriculosystemic shunt, extensive
craniotomy, high-grade histology, and the presence of a cerebellar lesion.
When organ donation is being considered, the local Organ Procurement
Organization (OPO) and the transplant team must consider all of this
information in the context of the patients awaiting transplantation. e
risk of transmission of a tumor from a donor with a primary brain tumor
is dicult to quantify but appears to be low, with identiable risk factors
that increase the risk. It has been recommended that potential organ
recipients be counseled around the small but denite risk of transmission
of malignancy, as well as the chance of survival if they choose to remain
on the waiting list for their needed organ. With donors considered to be
higher risk, transplant teams may exclusively request certain types of tissue
donation (for example, heart valves, cornea, bone, other), where the risk of
transmission is practically nil.
e Process of Organ and Tissue Donation
As parents of a child with a brain tumor, you may be considering organ and tissue
donation at different times during the course of your child’s illness. It is helpful
to speak with the health care team sooner rather than later in order to explore
options and the implications for your child’s care around the time of death. The
health care team will then refer your family to the local OPO and a representative
Chapter 20: Organ and Tissue Donation
288
289
Chapter 20: Organ and Tissue Donation
will meet with you to discuss the donation process.
Organ donation occurs following conrmation of a donor’s death with the goal
of maintaining the organs in a healthy state until the time of transplantation. e
process of removal of the organs from the donor is called organ retrieval. With
brain death, there is conrmation of irreversible brain damage but continued
heart activity, such that the organs are still perfused, or receiving their blood
supply from their donor. Donors are maintained on articial life support until
organ retrieval occurs. Some hospitals will oer donation after cardiac death,
after careful ethical consideration. In those situations, a donor is removed from
all life support and death is conrmed by lack of heartbeat and breathing eort.
Organ retrieval occurs immediately thereafter. In either case, death must occur
in the hospital for donation to occur.
Current guidelines recommend careful review of a donor’s medical history for
the risk factors discussed above, as well as careful exploration at the time of organ
retrieval to assess for metastatic disease. e areas to assess may include sites of
previous surgery, related lymph nodes, and the shunt tract, including the chest,
abdomen, and pelvis. In the unlikely event that spread of malignancy outside
of the brain is conrmed by pathology, the transplant team will be notied
immediately and the organs retrieved will likely not be used for transplantation.
e process around tissue donation depends on the tissues to be donated and
will require discussion with the OPO representative. For cornea donation for
example, the tissue is less sensitive to lack of oxygen and retrieval can take place
hours after death. In this situation, there is more exibility for families around
the time of death.
At the time of death
Every family will have their own needs and wishes for their child at the time of
death, and organ donation may not be appropriate or possible for many families.
For those families who do want to pursue the opportunity to donate, it is
important to understand the small possibility that your gift of organs may
be denied. (is is much less likely for gifts of tissue.) It does appear that
transplant teams now have better data to appreciate how small the risk of tumor
transmission actually is, and to counsel potential organ recipients appropriately.
ere is an urgent need for organ donors worldwide and patients with primary
brain tumors and their families have developed an extraordinary legacy with
their gifts of life.
Parent Perspectives
Parents do need the option of organ donation; therefore, they need to know
the right questions to ask and when to ask them. It's so difficult because
in asking the questions we are admitting that we are going to lose our
children—something we try to condition ourselves not to think about.
When we thought we were going to lose Emma I asked about organ donation
but was told we couldn't donate. Tissue donation was possible—hence the
corneal donation. The morning she earned her wings, our palliative care
medical team was connecting with the agency and we received the news
that they would accept Emma's corneas. It was so bittersweet to hear that
decision. Emma had an amazing way of seeing the world, people and events.
And now, there are two people that will be able to see their world, through
Emma's eyes. We take comfort in knowing this, and knowing that part of
Emma lives on in this world.
Soon after Mara's passing we received a phone call from the donation
center verifying our intent to have the corneas donated. We could have
missed this phone call had it not been for others answering phones for us
at the time. Just a few weeks later, we received a letter from the donation
center indicating to us that Mara's corneas were indeed used. We cried
tears of joy knowing that a small piece of our daughter was still alive. It
made us want to find the person and gaze into their eyes. It was made clear
to us through the letter that anonymity was paramount in this situation,
however. I understand why this has to be and appreciate that we can write
an anonymous letter to the recipient telling them about Mara.
I want to start by telling everyone a story about my family. My Mom lost her
husband, son, and mother all in a car accident on Mother's Day in 1963.
It was before I was born. My mom and my two sisters lived but the others
died. My brother's name was Max. Ironically, Max was 8 years old when
he passed away, the same age as my son Ethan. Max was killed instantly
and the one thing that my mother always said when I was growing up was
that she wishes she would have been able to donate Max's organs. That
Chapter 20: Organ and Tissue Donation
290
291
Chapter 20: Organ and Tissue Donation
even though it wouldn't have taken the pain away of losing him, it would
make her feel like his death had some purpose. And so when Ethan was
diagnosed with DIPG and his tumor later progressed, I felt a very strong
need to donate whatever organs, or cornea, or tumor that I could.
Like most of you, I always thought if you were a cancer patient you could
not donate your organs. In reading other children's websites I noticed that
many were donating their tumors and their corneas. So as Ethan's health
declined, I emailed his oncologist and asked him what we needed to do to
make pre-arrangements for this. At that point in time, he emailed me back
and said, “Lets cross that bridge when we get to it.”
A few weeks before Ethan passed away; we sat down with Ethan’s oncologist
and asked him again about tumor and cornea donation. He told me that as
Ethan's tumor is a glioma, they are known to not spread to other parts of
the body, i.e., the organs. He thought that Ethan may be able to donate all
of his major organs. He made some phone calls and put us in touch with
the Gift of Life Organization.
They told us that if Ethan was to be a candidate for donation, he would
have to pass away at the hospital. We had been on hospice with Ethan since
October so it would mean that the plans we had made for Ethan to pass
away peacefully at home would have to change if we wanted to donate. So
in talking with Ethan's father, he at first was not sure if he wanted to donate
but would think about it. I believed in leaving this to God, so decided that
if we were meant to donate Ethan's organs we would make it to a hospital
and he would die there. If we weren't, then he would pass away at home.
During Ethan's final days, he was in so much pain due to headaches. He
had never complained hardly ever of headaches previously, only very minor
ones. These were “over the top,” leaving him hollering in pain. Hospice
gave him morphine but it was not easing his pain. The hospice providers
kept in close touch with Ethan’s doctor and on Tuesday, he suggested we
should bring Ethan to the hospital to get his pain under control. So we
packed him up for the 1 hour 15 min ride to the hospital. The car ride up
there I rode in the back seat with him stroking his hair and forehead and
holding his hand.
When we got to the hospital, and laid him on the bed, I noticed his fingers
and toes were a bluish color. They hooked up the pulse oximeter and he only
had 10% oxygen saturation. So he was admitted to PICU and was eventually
put on a ventilator. A CT scan of his head showed that the tumor had grown
and was compressing areas of the brain that controlled breathing, but that
the brain had not herniated or hemorrhaged. The doctors told us that the
brain had been pushed upwards and that it was blocking the flow of spinal
fluid throughout the brain. This was something he could not recover from
and if taken off the ventilator he would not be able to breathe on his own.
But also that he probably would not have brain death for quite some time,
(days or weeks).
We were forced to make a decision: 1) continue to support him, knowing he
will not recover and that he is in discomfort; 2) make a decision to remove
support in his hospital room, and let him pass away as it would occur,
knowing he would not be able to donate any organs, or 3) plan to remove
life support in the operating room, with my husband and I present. They
would allow him to take his last breath and his heart to stop beating, and
then life support would be placed back on. At that point in time we would
have to leave the operating room so that the transplantable organs could
be removed (this is called a post-cardiac death transplant). If Ethan did
not pass away within a certain time frame of removing life support, then
he would be brought back to his room in the PICU, and we would be with
him until the very end, but not able to donate his organs.
There had only been four previous post-cardiac death organ donations done
at this hospital in 10 years. Ethan's case had to be brought to the Ethics
Committee to make sure it was ethical in his case. It was decided it was, so
we planned for him to be taken off the ventilator at 6:00 p.m. the following
evening in the operating room and donate his organs.
The Gift of Life Organization was able to locate one match for Ethan's
kidneys from someone who was willing to take them from a cancer patient,
and two recipients for his corneas. It was decided later that his tumor
would go to research.
So at 6:45 p.m. we were changed into scrubs and taken into the operating
room where Ethan was already prepped for surgery. They had all of the
surgical instruments covered up, and had Ethan draped with the exception
of his face and hands. His dad and stepmom stayed at one side and his
stepfather and I at the other. At 7:30 p.m. they removed the breathing tube
and we waited holding his hands for him to pass. He took several last
gasping breaths but then did not breathe again for several minutes even
though his heart was still beating. His heart would flatline but then register
Chapter 20: Organ and Tissue Donation
292
and then flatline and then register. Finally it stayed flatlined and the doctor
confirmed he had passed. We said our goodbyes and left the OR, while the
surgical team recovered the organs. Afterwards, they cleaned him up and
we were able to come and see him before leaving the hospital. He looked
very much at peace.
We had to give up the chance to have him at home for his death but I know
we made the right decision. I know in the end that Ethan would have wanted
his organs to save other lives, and I know he would have been proud of us
for making this difficult decision. It was evident when they removed the
tube that he could not breathe on his own enough to sustain his life. I think
his time to go was Tuesday evening, we just were fortunate to get two more
days with him while making preparations for his Gifts of Life. While it may
have changed where he died, we were able to see that he was at peace, and
know that someone else was able to live and see because of our sacrifice.

293
Chapter 21: Integrating Palliative Care
Chapter 21
Integrating Palliative
Care and Making
Difcult Decisions
Justin N. Baker, MD, FAAP, FAAHPM
Adam J. Tyson, MD
Javier R. Kane, MD
As you have read in prior chapters of this book, diuse intrinsic pontine glioma
(DIPG) is a serious disease. e majority of patients with DIPG and/or their
families are told that the tumor cannot be removed with surgery because of its
location. ey are also told that the tumor is rarely sensitive to chemotherapy,
and that while the tumor may decrease in size with radiation therapy, the disease
often continues to grow despite best eorts. What this means is that DIPG is
usually an incurable and progressive disease. When presented with such dicult
news, most families want their child to receive the best possible treatment for
this terrible disease, hoping that their child will beat the odds. Most families also
realize that serious illness often comes with pain and suering, and they want to
avoid this for their child as much as possible.
Parents in these circumstances often report that making decisions regarding their
child’s care is extremely dicult because it requires balancing the dual goals of
care that include cancer-directed goals and care that provides comfort-directed
goals. Parents often wonder how they can
possibly prepare for such an overwhelming
and dicult situation and how to plan for
their child’s care in such a way that hopes
for the best but allows for consideration
of all possible outcomes, thus allowing a
balanced approach to decision making.
In these dicult situations, parents must
often think about what is most important
Dr. Baker is the Director of the
Quality of Life and Palliative
Care Division, Director of
the Hematology/Oncology
Fellowship Program, and an
Attending Physician for the
Quality of Life Service at St.
Jude Children’s Research
Hospital, Memphis, TN.

Chapter 21: Integrating Palliative Care
294
295
Chapter 21: Integrating Palliative Care
for the child and family. Based on what is happening with the tumor, available
medical treatment, and the child’s experience, the parents must decide the most
important goals in the care of their child. Families may also have important life
goals they would like to accomplish with their child. ese goals are critical and
should be taken as seriously as evaluating blood levels or assessing the side eects
of medications. Every attempt should be made to integrate these goals into the
overall plan of care for children with DIPG so as to balance the use of treatments
to ght the tumor, and the eorts of the family and the care team to preserve
comfort and the best quality of life (QOL) possible for these children.
Establishing Goals of Care and Making Dicult Decisions
Most families start treatment with the hope of attaining a cure for their child with
DIPG. Having hope is very important, as it sustains patients, families, and their
caregivers during extremely dicult times. Unfortunately, this disease makes cure
highly unlikely and in reality most therapies for DIPG are either experimental or are
only intended to prolong life. is does not mean the team will not remain hopeful
with you that a cure can be attained, but it does mean that you and your team must
hope for the best possible results while planning for all possibilities, including the
possibility that the tumor will continue to grow in size despite everyone’s best eorts.
If and when the tumor begins to grow despite the treatment, parents may know with
greater certainty that the child’s tumor is incurable. During this time it is appropriate
to continue to hope for the best possible outcome. If you come to realize that your
child has a disease that cannot be cured, it is also critical to work on deciding the
goals you would like to try to achieve while your child is alive.
You may want to think about what is most important to you and your family,
and how you would like to spend the remaining time you have with your child.
You will once again face many dicult decisions. It may be easier for you to make
decisions at this time if you think about whether or not the possible treatment
choices presented to you will help you achieve the goals you have for your child
and family. Parents report that the most dicult decisions they have to make
for their child happen at this stage and include whether or not to stop ghting
the tumor and stop cancer treatment or to
enroll their child in a phase I research trial
[Table: 1].
Goal to prolong a life of good quality—minimal morbidity
Values discussion regarding location of care and cancer-directed
treatment options:
• Phase I trial possibilities
• IV or oral cytotoxic chemotherapy with hopes of tumor response
• "Palliative" chemotherapy
Values discussion regarding priorities and life plan goals:
• Discussion with your child about DIPG progression
• Hospice enrollment
• Placement of a DNAR order
Goal to optimize comfort
Values discussion regarding priorities and life plan goals:
• Discussion with your child about progression
• Hospice enrollment
• Placement of a DNAR order
Table 1: Goals and values discussion
Parents may also choose to avoid further pain and suering caused by invasive
treatments such as breathing machines or other aggressive and potentially
uncomfortable life-sustaining treatments and have a “Do Not Attempt
Resuscitation” (DNAR) order (see questions 6 to 8 in addendum below for
more information about DNAR orders) placed in the medical record, or to
decide against certain treatments that may not help achieve the primary goals
of care. Other important decisions to consider include whether or not to enroll
their child in hospice, to speak to their child about the fact that the disease
is worsening or the possibility of death, choosing a desired location of death,
deciding whether an autopsy should be performed, and other important life
choices such as whether or not to continue going to school, traveling, or pursuing
other goals the child and family would like to achieve. While these are some
of the most dicult decisions you will ever
have to make, the specic goals of care you
have set for your child and family will help
minimize distress for your child and help
Dr. Kane is the Director,
Pediatric Hematology/Oncology
and Professor in the Department
of Pediatrics at The Children’s
Hospital at Scott and White,
and Texas A&M Health Science
Center, Temple, TX.
Dr. Tyson is a Family Medicine
Resident Physician at the Bryn
Mawr Hospital, Bryn Mawr, PA.

Chapter 21: Integrating Palliative Care
296
297
Chapter 21: Integrating Palliative Care
to ensure his or her comfort.
Your Team and Individualized Care
You and your child are part of a team. In fact, you are the most important members
of this team. Your health care team is taking this journey with you, and everyone’s
desire is that you and your child should never feel alone. Your care team may not
be able to fully put themselves in your shoes, but they have likely walked through
this very dicult time with many other families and will draw on those experiences
to help you. You should encourage your medical team to be very open with their
thinking as they make recommendations to help accomplish the various goals of
care that you have for your child and family. is process may look dierent for
your family than it does for others [Fig. 1]. Discussing the individualized aspects
of care is dicult, but the better the team knows you and your child, the better
the care plan can be matched to your and your child’s needs.
Figure 1: Decision-making schematic
Most children with DIPG enroll in a research treatment plan or receive treatment
following a specic cancer treatment plan. You may be wondering how this
individualized approach to palliative care can work within the context of a research
protocol or a predesigned cancer treatment plan. Remember that these treatments
are only one aspect of the overall care plan. A care plan includes a medical plan
and a life plan, both of which should be individualized to your and your child’s
unique needs. A specic research protocol or cancer treatment plan may be a part
of the medical plan, but other aspects of the care plan can be highly individualized,
such as medications to relieve pain and other distressing symptoms. Any specic
needs that are unique to your situation, such as rehabilitation needs or spiritual
needs must also be addressed.
Communicating your needs and perspectives
Communication with your child’s physician and other members of the care team
about all of the before-mentioned issues is extremely important. As your team
(remember this means your child, family, and medical care team) starts out on
this journey together, you can facilitate better communication and understanding
by talking to the medical team as much about your child and family as possible.
Do not hesitate to bring your child’s and family’s needs to the attention of your
care team, because they cannot address your needs if they do not know what
your concerns are or what is most important to you. e better the team knows
you and your child, the better the team can make an individualized plan of care
along with you.
You may not automatically know what information is important to share. It will
be helpful for the team to be able to judge your level of understanding of the
prognosis, goals, and treatment options as treatment begins, and at other key
points in time if the illness progresses. It will also be very helpful, for example,
to hear about whom your child is, how your family is doing, and what the illness
experience has been like for each of you.
Questions to consider when talking to the team about these things are listed in
Table 2.
Category Questions to Consider
Understanding your perspective What does good quality of life mean
for you and your child?
What are you hoping for? What is
your child hoping for?
What is most important for you and
your family?
What are you most concerned about?
What is your denition of being a
good parent to your child?

Chapter 21: Integrating Palliative Care
298
299
Chapter 21: Integrating Palliative Care
Category Questions to Consider
Information and decision making What information do you or your
child need right now?
How do you like information to be
delivered/handled?
How much does your child want to
participate in decision making and
dicult conversations?
Specic needs: symptoms What are your child’s most concerning
or distressing symptoms that interfere
with good quality of life?
Specic needs: spiritual Are you or your child experiencing
spiritual distress? Do you feel aban-
doned by God? Do you feel angry
toward God?
Specic needs: emotional Is anyone in the family experiencing
emotional distress that is interfering
with good quality of life?
Specic needs: social Are there any family needs that if
not addressed will lead to increased
distress?
Are there any sibling needs that are
currently not being addressed?
Chance for treatment success What is your family’s understanding
of your child’s chance for cure and
overall life expectancy?
Goals What are your goals for treatment?
What other goals do you have for your
family and your child?
Treatment alternatives What is your understanding of the
availability of cancer-directed treat-
ment options?
Table 2: Helpful perspectives for your child’s health care providers
Reconsider these questions from Table 2 periodically and tell the rest of the care
team of any changes. e diagnosis of a DIPG is overwhelming for all families,
and the way you look at the tumor and its impact on your child and family
is likely to change as time goes by and as symptoms resolve and/or progress.
Some key moments when most families should consider these questions include:
• at diagnosis and the beginning of treatment;
• at the end of radiation therapy;
• upon returning for the rst magnetic resonance imaging (MRI) scan
following radiation;
• when symptoms seem to be returning;
• upon conrmation that the tumor has progressed on imaging;
• as your child’s condition declines or the symptoms continue to progress,
or worsen.
Your family’s level of understanding is likely to shift during any and all of
these events and your goals of care for your child and family are also likely to
change. e medical team will not be aware of these changes unless you help
them understand your evolving thoughts and goals.
e key to establishing goals of care is open communication between you, your
child, and the primary medical team.
Some examples of goals of care are noted here:
• Cure of disease;
• Prolonging life, while having the best quality of life possible;
• Providing comfort;
• Maintaining or improving the ability to perform activities of daily
living;
• Attaining specific life goals (e.g., going to graduation, camp, wish
trip);
• Support for family and loved ones;
• Advancing medical knowledge (i.e., helping contribute to a cure);
• “Knowing we did all we could” (i.e., that we did not give up);
• “Being the best parent that I can be” (i.e., making the best decisions for
my child).
ese goals are not necessarily mutually exclusive. Many families will choose
dierent goals of care at dierent points in time, and the list above is only a
Chapter 21: Integrating Palliative Care
300
301
Chapter 21: Integrating Palliative Care
sample of the large number of possibilities. Table 3 contains decisions you may
need to make for your child, along with possible positive and negative aspects
of these decisions.
Decision Potential Positive
Aspects
Potential Negative
Aspects
Further cancer-directed
therapy.
• Slow progression of
the tumor.
• Fulllls a need to
continue to ght
against the tumor.
• Introduces or in-
creases suering due
to side eects.
• Continued need for
medical care primar-
ily provided through
outpatient clinic or
hospital setting, so
less time to pursue
other life goals.
Enrollment in a phase I
study
• Further understand-
ing of how the
medicine works.
• Involvement in re-
search altruism; a
chance to give back.
• Closer monitoring
of clinical status.
• Fullls a need to
continue to ght
against the tumor.
• No studies may be
available.
• Introduces or in-
creases suering due
to side eects.
• Continued need for
medical care primar-
ily provided through
outpatient clinic or
hospital setting, so
less time to pursue
other life goals.
Decision Potential Positive
Aspects
Potential Negative
Aspects
Hospice enrollment • Home-based provi-
sion of care.
• Expertise in pain
and symptom man-
agement.
• Interdisciplinary
team approach to
care—availability of
chaplain, physician,
nurse, social worker,
and volunteers.
• 24/7 call coverage
for symptom-related
or other emergen-
cies.
• May be viewed by
others as “giving
up,” because hospice
is sometimes viewed
by the general
public as being for
people dying soon.
• Another team work-
ing with you and
your child; meeting
new people may
seem dicult.
• May not allow for
blood product trans-
fusions or continu-
ing cancer-directed
therapies.
Placement of a DNAR
order
• Tells all team mem-
bers that benets of
aggressive resusci-
tation eorts are
outweighed by the
suering such ef-
forts may inict.
• Focuses the nal
moments on com-
fort and mourning.
• Can be changed/re-
versed at any point
in time.
• May be viewed as
“giving up,” or not
doing everything
possible.
• Because it is a piece
of paper, it may not
be followed unless
presented to medical
personnel when the
patient is actively
dying.
• Can be a statement
of acceptance that
the end result will
likely be death.

Chapter 21: Integrating Palliative Care
302
303
Chapter 21: Integrating Palliative Care
Decision Potential Positive
Aspects
Potential Negative
Aspects
Aggressive symptom
control
• Decreasing symp-
toms allows one
to focus on other
important aspects of
life and the things
that can be enjoyed.
• Helps decrease suf-
fering.
• May cause drowsi-
ness or sleep.
• Certain pain
medications may
cause unintended
consequences (e.g.,
signicant constipa-
tion) if not treated
properly.
Location of death: home • Allows the child
to be in a familiar
environment.
• Limited interrup-
tions (e.g., no nurse
taking vitals con-
stantly, noisy alarms
going o, doctors
being paged over-
head).
• Ease of access for
friends and family
(no visitor hours).
• May decrease costs.
• May feel less sup-
ported (i.e., we tend
to look to hospitals
or clinics for medi-
cal support instead
of having it deliv-
ered in the home).
• Limited gatekeepers
(e.g., hospital sta
and nurses who can
help control who
has access to family/
patient).
Location of death:
hospital
• May allow sta to
help control visitors
(keeping visits to a
minimum).
• Continual access
to nursing sta and
physicians at the
bedside.
• Noisy with many
interruptions.
• An unfamiliar envi-
ronment.
• Limited and con-
ned space.
• May increase costs.
Decision Potential Positive
Aspects
Potential Negative
Aspects
Speaking to my child
about death and dying
• Allows you to ad-
dress/relieve the
fears and concerns
your child may not
express without be-
ing invited to share.
• May provide a sense
of closeness and un-
derstanding between
you and your child
during this process.
• May relieve your
child to know that
you will be okay and
will be able to cope.
• Dicult emotion-
ally.
• Takes time.
• Requires facing the
fears of saying “the
wrong thing.”
Table 3: Specic decisions, along with positive and negative aspects of each decision
Pediatric Palliative Care
Children with DIPG often benet from palliative care. Palliative care is specialized
health care designed to relieve suering and provide the best possible quality of life
for people facing the pain, symptoms, and stresses of serious illness. If your child has
DIPG you may decide that having access to palliative care resources is the right option.
Palliative care aims to promote healing and increase the quality of life throughout
a child and family’s journey through serious illness. e aims of pediatric palliative
care are not limited to a disease process (e.g., DIPG), but rather become helpful
for improving quality of life, maintaining dignity, and attending to the suering
of seriously ill or dying children in ways that are appropriate to their upbringing,
culture, and community.
Pediatric palliative care promotes a team approach to care focused on addressing the
patient’s and family’s needs and on providing the highest quality of care. is care
should help with dicult decision making and care planning; attend to suering
from physical or psychological symptoms; address social, emotional, and spiritual
needs; improve communication and coordination of services; be a point of continuity
no matter where the child is receiving care; provide the highest quality hospice and
end-of-life care; and address grief and bereavement issues.
Chapter 21: Integrating Palliative Care
304
305
Chapter 21: Integrating Palliative Care
Hence, palliative care is about giving the child the best quality of life possible,
regardless of whether or not the child is receiving cancer treatment. In this regard,
a child with DIPG may have access to palliative care services provided by members
of the primary oncology care team while he or she receives cancer-directed therapy.
Adding pediatric palliative care resources and principles into the overall plan of
care helps the primary care team and your child and family work together, with the
ultimate goal of providing the best possible treatment for the disease while creating
the most comfort and best quality of life. In your hospital, your primary care team
may also be able to consult with a specialized pediatric palliative care team to help
you and your child achieve the best quality of life possible.
Addendum
e following section provides some specic examples of some frequently asked
questions and possible decisions.
1. What are some of the things you want to consider in making dicult decisions?
Every decision and situation is unique. However, some things that parents think
about include:
• What your child wants;
• Care team recommendations;
• Your personal feelings, such as how to be a good parent;
• Faith/beliefs;
• How the decision aects the family.
Some reasons children give for making the dicult choices they make include:
• Avoiding treatment that will make them feel worse;
• Specic life goals (e.g., going to graduation, prom, camp, wish trip);
• Pursuing comfort when cure is no longer an option;
• Pursuing specic care-directed goals, including not wanting to continue therapy;
• Seeing how other patients around them have suered.
2. Who can help me make these decisions?
Your child’s primary medical team is generally looked to for the most support. Many
families also ask for help from other health care sta, family, friends, and spiritual
leaders. Also, other families you have come to know through the DIPG journey
with your child may have helpful insights. It will be important to recognize that
these decisions are understandably very dicult, and because they are so dicult,
it is important to realize when you need help making them. You may need help
in getting information or talking with others who have more experience with this
process. Any time you realize that you need help making these decisions, ask your
clinical team for assistance. Ongoing communication is crucial in every aspect of
this process. Recalling your specic goals of care will be helpful in directing these
decisions. Once you have identied those goals, share them with your team regularly
so they can maintain your goals as they suggest a course of treatment for your child.
Depending on your child’s age and situation, it may be appropriate to discuss these
decisions with your child. He or she may bring insight and revelation that the team
can draw on when making decisions. If you have doubts about whether or not to
discuss these decisions with your child, ask for help in determining whether or not
it would be right or helpful.
3. What kinds of decisions will I be making?
Some decisions, in general terms, are centered on whether or not to:
• Enroll in phase I or II experimental clinical therapies, which do not have a
curative goal but may oer more quality time for the patient;
• Try treatment to prolong life (e.g., radiation, chemotherapy, surgery);
• Pursue aggressive symptom control for improved quality of life;
• Create a DNAR order for your child;
• Choose the location of your child’s death;
• Speak with your child about death and dying.
4. How can I help the medical team help my child?
Many times the treatment decision and plan are based on specic goals of care as
established by you and your child. ese goals may be as simple as “aggressively treat
pain,” or as complex as “I want my child to be able to perform normal activities for
a child his/her age.” Presenting these goals clearly to the medical sta needs to be a
top priority. ese goals should be agreed upon by you, your child, and your child’s
primary care team. From these goals, a particular care plan can be established.
e medical sta will never know your child as well as you do. You must continue
to be his or her strongest advocate. is cannot be over-emphasized. You are very
Chapter 21: Integrating Palliative Care
306
307
Chapter 21: Integrating Palliative Care
likely to be more in tune with your child’s needs than anyone else, so you must be
his or her advocate. Any time you think there is something dierent the team should
be doing, share it with them. Any time you think your child is suering or could be
doing better, tell the team. Ongoing communication is the key.
5. Where should my child receive the remainder of his or her care?
Your child will continue to receive medical care to ght against pain and other
symptoms, no matter where you decide to receive care. Choosing to continue
receiving cancer-directed therapies on a study may limit your location options (e.g.,
your ability to return home or be close to home). is must be considered in the
context of overall goals of care. If being at home is important to your child and family,
plan ahead and organize a team of providers who are your best allies—work toward
this transition before an emergency or crisis arises that might hinder or completely
prevent you from going home (e.g., your child being in the intensive care unit on
a ventilator). Every eort will be made to help coordinate your child’s and family’s
goals of care throughout the treatment course.
6. What is a “Do Not Attempt Resuscitation Order” (DNAR) and should I have
my doctor place a DNAR order on my child’s chart?
A DNAR order is a request to allow a natural death for your child rather than
performing cardiopulmonary resuscitation (CPR) if your child’s heart stops or if he
or she stops breathing. You and your child’s primary medical team will consider such
an order in light of the goals of care for your child. It is best to make this decision
when the goals of care are changing, rather than during a time of crisis. If you decide
a DNAR is best for your child, the order is put in your child’s medical record by your
primary medical team. A DNAR order is most appropriate when using medicines,
procedures, or machines to restart your child’s heart or breathing are unlikely to
benet your child or when such measures may actually be harmful. is decision is
usually the case when the DIPG has started to grow again and your child’s physical
decline is due to the growth of the tumor.
As patients go home to receive care, many of them have an out-of-hospital DNAR
in place. It is best to plan for these events in advance, because unless given other
instructions, medical sta (i.e., hospital sta or emergency medical sta) will attempt
to resuscitate all patients whose hearts have stopped or who have stopped breathing.
7. Can I change my mind about the DNAR order for my child?
Yes, you can change your mind about the DNAR order. Feel free to discuss changing
or canceling the DNAR order with hospital or medical sta at any time. ese
decisions should again be considered in view of the goals of care for your child and
can be reversed at any point in time. Children are treated on a case-by-case basis,
and all types of treatment can be given, whether or not a DNAR order is in place.
e condition of your child and the agreed upon goals of care will determine the
clinical decision making for your child.
8. is is a very dicult decision; how do most parents decide?
Most children who have died from DIPG had a DNAR order in place at the time
of their death. Some parents know early on in treatment what they will decide about
a DNAR; others prefer to speak with their family, friends, their child’s pediatrician,
other parents of children with cancer, or their ill child, when possible. is is a very
dicult decision and it should be made with the goals of care for your child in mind
and with the help of your primary medical team. Your primary team can help you
look at all the options from dierent perspectives to help you make the best decision
with/for your child and your family.
9. How much do I tell my child?
Every situation is dierent and the level of understanding for children varies greatly.
ere is no right or wrong answer to this question. You know your child best, and
over the course of your child’s illness you have learned how much your child wants
to know and how your child handles information. Children as young as 9 or 10
years old, or even younger in some cases, understand complex situations and can
help in making dicult end-of-life decisions. Some families regret not speaking to
their child about these issues, especially if they feel their child knows or wants to
discuss such topics further. Studies have shown that parents who have discussed
death and dying with their child do not report any regrets about doing so. On the
other hand, some parents who did not discuss these issues with their children report
wishing they had done so.
10. What if I change my mind about the decision I have already made?
As time goes by it may be very appropriate to change your mind, or it may be
important to stay the course. If you want to change your decision or shift the focus of
care, the medical sta will do their best to support that decision. e rst thing that
will likely occur is to suggest a revisiting of the goals of care. From these goals, you
can decide as a team the best way to proceed. It is always important to consider all
aspects of these decisions and ask if these decisions will lead to more harm than good.
11. What if I want to pursue "complementary and alternative medicine" (CAM) or
other non-standard therapies such as herbs, nutritional and vitamin supplements,
acupuncture etc.?
Chapter 21: Integrating Palliative Care
308
309
Chapter 21: Integrating Palliative Care
Parent Perspectives
The following, are stories written by parents of children with DIPG. Some
of the stories may be emotionally difficult to read. As editor, I felt they were
important to include in the book in order to help you gain what I hope will
be a balance of perspectives, through the experiences of other parents who
have endured this stage of the journey. My intent is to provide you with
information that you can discuss ahead of time with your health care team,
and assist you with making decisions that are appropiate for your child.
Ruth I. Hoffman, MPH
No one's path with DIPG is the same, and the only thing you can strive for
is that you don't regret any of the decisions you've made.
We have been extremely lucky to be connected to an excellent palliative
care doctor who visits us weekly at home and gives us an incredible amount
of information and support. Although Stella is not receiving treatment,
she receives morphine on a daily basis to eliminate the pain in her head,
PEG flakes to counteract constipation, Zofran for nausea, and Ativan for
seizures. So far we have been able to give her all her medications orally…
mostly hidden in ice cream!
When we decided against treatment, doctors told us to expect Stella to live
3 to 4 months maximum. We are 8 months into the diagnosis, and Stella is
still with us. She has seen her brother being born, lived through Halloween,
Christmas, New Year's, Valentine's Day, etc. She still smiles on a daily
basis, and although she is declining, the decline is slow and often stalled.
We have been open and honest with Andrew about his situation.
Conversations regarding dying and Heaven have become fairly common
over the past couple of months. When you are dealing with cancer, especially
one with such a dismal prognosis, there is already a sense of isolation. We
have not lost hope, but we do understand the reality of what we are facing.
And we do not want to isolate Andrew further by making him feel that he
Talk openly with your child’s doctor and care team about this option. Often these
therapies have not been studied for their potential benet or harm to your child. If
the therapy is not seen as harmful, and you feel strongly about pursuing a particular
option(s), the team can try to watch for possible side eects and cross reactions with
other medications your child may be receiving.
Chapter 21: Integrating Palliative Care
310
311
Chapter 21: Integrating Palliative Care
cannot talk to his own family about the things that are on his heart. When a
child—or anyone—expresses his feelings, we do not help by making light of
those feelings or attempting to explain away those feelings. When Andrew says
he doesn’t want to die, we can honestly respond, “We don’t want you to die.”
Bryce was told that first week that other children who had had this kind
of tumor had not survived. My husband and I decided at this point that
if we were going to ask him to go through all of this treatment, then we
were going to have to be hopeful, even knowing what we knew, and after
researching on my own, I knew that the doctors were right. We decided
that we were going to follow Bryce’s lead, and so we asked his care team
to not talk about dying until Bryce wanted to know about it. That meant
informing every doctor who saw him that we were choosing this path before
they spoke to Bryce. How could we ask him to do all of these treatments
and still know that he would probably not survive?
It wasn’t until the end of June that he finally asked us about his future.
Children do things in such an uncanny way. He and I were at the gas station
when he finally asked. (We had some of our best talks in the car.) He said,
“Mom, what is going to happen now?” I asked what he meant, even though
I already knew where he was going with it. “Now that I’m done treatment.
What if it comes back?” So, at that point I reminded him about what we
had been told—that radiation could not be done in the same spot for a few
years; that they did not have any chemotherapy that was working for this
kind of tumor; and that surgery was not an option either. With tears in his
eyes, he responded with, “You mean they are just going to let me die?”
My response was, “I hope not Bryce. We will have to see if something new
comes along, but for whatever comes our way, we are going to just try to
enjoy every single day.” Next, he said, “Mom, go pump the gas.”
As Bryce began to feel worse, it was then that we started to talk about
dying. It would often be that Bryce chose to speak to me about it. He began
asking questions about things like: Would he be here for his birthday in
December? Would he be here for Christmas? Would he be here for his
cousin’s birthday next summer? My response would always be, “I hope
so Bryce.” I tried to be as honest as possible, and also let him know how
much I loved him at the same time.
He continued to go to school right up until December 19th. And on
December 20th he fell at home. We contacted the hospital because over
the weekend we were seeing a decline in his speech, abilities, and mental
capacity. An MRI was scheduled for Dec. 23rd. His oncologist called with
the results on December 24th to confirm what we already knew. The tumor
had progressed. At that time, she indicated that once the tumor progressed,
that it would more than likely be swift. So she referred Bryce to hospice
care and we signed papers for Care at Home. Life at that point felt like
it was spiraling out of control for all of us. Over the Christmas holidays,
Bryce progressed from walking on his own, to a walker, to a wheelchair, to
spending most of his time in bed as his ability to walk and move declined.
We decided at that point that our daughter would only go to school for half
a day. She needed the normalcy of going to school, but we felt that she also
needed some time at home with Bryce, without visitors. It was one of the
best things that we did, and it allowed her to feel like she was part of his
daily care. She would often crawl into bed with him and we would hear
them talking away. This was a stressful time for her too, and she was full
of questions about what was happening to him. We tried not to give her too
much information at once, or to let her know more than she needed to hear
at the time, because we felt that it would frighten her. But we always felt
that we needed to answer her questions clearly and honestly. We watched
her grow up in the process of watching her brother’s decline and throughout
his palliative care. It was almost as if she felt that she became the older
sibling throughout the process. She knew as much as we did by the end
because she even felt comfortable asking the nurses who came into our
house what was happening. I look back, and now feel that this is honestly
how she learned to cope with losing him too.
Talking about dying would pop up at the oddest moments, and catch me
off guard. I would make a point of stopping whatever I was doing because
I was afraid that he would not ask again, and I wanted him to have the
answers. One day he asked if his head was going to just explode. It became
very important then that we discuss what was going to happen medically.
As Bryce became unable to walk, the questions changed to what happens
after one dies and whether or not he would still be sick. He asked, “Will I
be able to walk in heaven?” I took this as some kind of acceptance of his
fate, and tried to follow his lead.
In reflection, we did make one mistake. One day, when Bryce referred to
the day when his head would explode because the tumor would not have
Chapter 21: Integrating Palliative Care
312
313
Chapter 21: Integrating Palliative Care
anywhere else to grow, it became very important to explain medically
about what would happen to him. None of us had really asked the BIG
question—we were afraid of the answer. I mentioned to him that eventually
he would just become tired and that his body would begin to shut down
and that that would be when he would be dying. He began having trouble
sleeping. We finally figured out, with help from our palliative care team,
that he was afraid to sleep because he thought that he was going to die in
his sleep. Obviously not a good choice of words to explain what was going
to happen to him.
One of the most difficult things about watching Bryce go through this was
that as his tumor progressed, he became more anxious, and had more pain.
As this happened, Bryce began talking of dying more freely. Every morning,
because his mental capacity and memory became affected, he would wake
up and yell out “I’m scared!” We would ask him what he was scared about.
He would say “I don’t remember what you told me about heaven!” So we
would spend time every morning reminding him of what our family believes
happens when someone dies. And we began having to give him Ativan for
his anxiety. He then began asking us to put his hands together every night,
so that he could say his prayers.
About two weeks before Bryce’s passing, he was in pain and very anxious
and he actually looked at me, and asked me if I would kill him. I thought
I was going to die myself with heartbreak. But I looked at him, held his
hands very tightly, and said “Bryce, I know that you are frustrated, and
angry with what is happening to you, and that you can’t find the right words
to tell us how scary and awful this is. When you are ready to go because
you are tired of fighting, I want you to know that it’s okay. I love you with
every breath that I take and every beat of my heart, but I can’t kill you—it’s
illegal.” And he laughed, but I knew that he was serious. I feel that that was
the day that he honestly tried to show us that he accepted what was coming.
Talk about dying changed into making us promise that he would be buried
near his grandparents or by the pond at the cemetery, and into making us
promise to go there to visit him every day. He was afraid of being forgotten,
which is apparently normal for most teenagers.
I had been diagnosed with colon cancer in late June and was scheduled to
have the cancer resected on July 13th. So, the plan was that I would have
surgery in the morning and as soon as I came out of recovery, my husband
and Caleb would drive to Memphis so Caleb could start treatment the next
day. Caleb snuggled up in bed with me for a while before they hit the road
and then they were off.
Caleb began declining rapidly during the drive. By the time they got to
Memphis at about midnight, my husband was worried that Caleb wouldn't
be able to qualify for the study. He was declining quickly. By the next
morning, the only concern became whether they'd be able make the hour
trip home before he died. He was declining quickly and obviously could
not be treated. My husband and I discussed it and decided we would take
the risk. We wanted him to be home.
As we left the hospital, Caleb was in a wheelchair. His breathing was
more labored and he'd just been placed on oxygen that day. We arrived
home and the hospice nurse and equipment was there and ready to be put
in place. The guy who brought the hospital bed had quite a time getting it
put together. He made the comment that it was an old bed and so heavy. He
had no idea why he'd been required to bring it for Caleb. Our nurse told
us that she has a 10 year old son and she knew that if her son was dying
at home, the whole entire family would want to be piled in bed with him.
She wanted to make sure to get us the strongest and biggest bed she could
so that we could all pile in whenever we wanted. I carry that memory with
me as an example of the blessings so many people gave us along the way.
So, Caleb was set up in the living room. We had an open door policy.
Friends and family were welcome to come and stay as long as they liked.
Some of our closest friends were with us the entire time. Some of Caleb's
closest friends came and just sat by his bed for hours. By this time, Caleb
could only communicate by moving his eyes up and down. Even the side-
to-side movement was gone. I will never forget that I was sitting on his
bed, talking with his best friend Samuel, and Caleb managed to make a
joke by moving his eyes up and down during our conversation! Caleb's 5th
grade teacher came by to check on him often, and we asked her to contact
any of his classmates who would like to spend some time with him and let
them know they were welcome to come and visit. So, we had many children
come through our home to spend time with Caleb over the next two days.
By the following morning, Caleb was not very responsive and did not really
interact with anyone. The home health aide came mid-morning to check
on him and she and my husband gave Caleb a bath. My husband noticed
Chapter 21: Integrating Palliative Care
314
315
Chapter 21: Integrating Palliative Care
that Caleb’s breathing changed and knew the end was near. He called our
other boys in to be with Caleb and the friends who were here went into
another room. Caleb peacefully took his last breath with his brothers and
daddy holding him in their arms.
Our hospice care was subpar, at best. We felt that our highly recommended
hospice staff was not adequately trained to deal with children, especially
children with this type of brain tumor. We also felt the staff was lacking
in compassion, the ability to read parents, and deliver the proper care to
our son. Sadly, we did not get the privilege of time to adequately research
hospice care, so we trusted our medical staff and their recommendation.
I wish I could say our Connor passed away peacefully, but after my many
urges that he was still suffering, I was simply dismissed. The end was even
more horrific and it really hurts to talk about it. We had a hospice nurse
trying to get an oxygen tank for his much labored breathing (which we
had been requesting for days). After suffering for a long time, he died.
Our hospice nurse, who was on the phone at the time trying to secure an
oxygen tank, simply said, in a calm manner, "No, never mind, we no longer
need it…the patient has expired." Really? My sweet, precious little boy has
"expired" to you? I will never forget those final moments and have to say
the hospice staff was unbelievably unprofessional and lacked compassion
in our situation.
It has been 22 days since my son's passing and we still haven't had a
response from a hospice bereavement counselor.
When I had time, I would send an email or call a friend telling them about
the conversations I was having with my kids. I didn’t know if I was saying the
right thing and I wanted a counselor to help me navigate these land mines.
I have a couple of friends in the counseling profession so I knew I could
trust their perspective. St. Jude offered professional counseling through
the child life specialist or the social worker if I needed more support for
myself or my kids.
I learned that giving my kids the opportunity to talk about what was in
their heart gave them the freedom to heal and move in and out of the stages
of grief just like I moved through these stages. Children have immature
ways of handling emotions and if I can help my kids by listening without
criticizing or interrupting and asking questions, then it makes them feel
loved and safe, and they are able to handle their grief until a professional
counseling environment can be offered.
Scan results confirmed our growing fears. Julian’s doctors braced us with
the news that the “relentless” disease had taken over and that we had less
than a month left with our sweet boy. Turns out, we only had five days.
Barely five years old, we spared Julian unnecessary fear and anxiety, and
never really explained that we would soon have to say good-bye. Wonderful
hospice nurses and doctors coached us on comfort care and what to expect
as the end drew near. We, intent on keeping Julian happy and at ease,
surrounded him with his favorite toys, books, movies, music and most
importantly, his favorite people.
In his final moments, we assured him over and over that it was ok to go, that
he was safe and loved and that we would be ok too. I held him in my arms,
and my husband held us both in his, and together we ushered our child out
of this world, just as we had ushered him in. Julian, ever the generous of
heart, left us with one final gift just moments after he passed. He wore on
his beautiful face the most serene, almost blissful expression, in which he
seemed to be telling us, “We did it. I’m in a wonderful place now. And you
guys aren’t even going to believe how good this gets!”
Liam passed away at home in my arms with his daddy and siblings
whispering their “I love You's” in his ear, and with his extended family in
the rooms nearby. Nearly 14 months after his diagnosis he finally won his
peace. He is a magical boy and dearly, dearly missed.
Mara passed away after a 12-hour intense struggle, at 8:12 a.m. on
September 22nd. Both sets of grandparents, 2 aunts, her siblings and
Bryan and I were all nearby when Mara slipped away. Our hospice nurse
arrived very soon after Mara’s passing (Hospice nurses had been in and
out throughout the course of the night) and started making preparations.
I began the process of getting my little girl ready for nearly the last time.
Chapter 21: Integrating Palliative Care
316
317
Chapter 21: Integrating Palliative Care
When the moment was right (probably an hour after she passed) I began
to bathe her, dress her, and do her hair. Grandma helped Natalie (Mara's
younger sister) pick out the right nail polish for her fingernails. We put
on just the right dress and picked out her favorite jewelry. We took some
precious pictures of the kids' hands together. This time with her was so
very painful, yet it was peaceful. It was excruciating but sacred. I would
not have traded this precious time for anything. The funeral home came at
approximately 10:00 a.m. to pick up Mara. We held a family prayer and
then Bryan carried his baby girl for the last time to the van (rather than
use a stretcher) in a final act of love and compassion.
Looking back, there are moments when I wished I had perhaps one more
hour with Mara's body before they took her away. However, things were
beginning to happen to her body (blood was starting to pool—something
we were told is very normal) and I'm not sure that I'd like to witness every
detail of nature taking its course. We did everything we could have done.
We did have the privilege of dressing her at the funeral home 4 days later
before her viewing. It was nice knowing that we'd have one more chance
to see her alone. Sure, it was not the same but in some ways Mara looked
more beautiful at the funeral home than she did at our home. I don't think
as a mom that I realized how sick Mara truly was. That probably sounds
silly, how could I not know, right? But I just think that we kept picking up
and moving forward so many times that I didn't absorb completely what
was happening. It wasn't until I looked back at pictures of her last days
that I could see how sick she was. At the time, I just saw my perfect angel
without any imperfections. Even to this day when I think of memories with
Mara, I think of her running and playing…not lying on the couch with a
feeding tube in her nose.
Our oncologist was brutally honest, which we appreciated to no end. But
even when things were stable, they kept reminding us, "It will come back;
it’s just a matter of time." We wanted to celebrate the small things, even if it
was just for a couple of hours, but were usually crushed before leaving the
hospital room. This is after getting “good" news, i.e. stable tumor. We all
know it is "coming back," but do the doctors really need to constantly drill
it in? Trust me no parent EVER forgets that this tumor usually comes back.
Imagine that you had a cherubic, mischievous, energetic and moody two
year old with flashing blue eyes, a brilliant smile and curly red hair.
Imagine that each morning she got you up at 5:15 am by standing up in
her crib and shouting, "Maaamaaa, I'm awaaaake! Maaamaaa, where are
you??" Imagine if when you went into her room she threw both her arms up
towards you in a great big hug and chattered her way into the living room,
telling you she wanted Cheerios for breakfast…with banana…and milk…
and when is Auntie Heather coming…and can we paint now…and watch
Caillou. Imagine if when you tried to get her dressed in the morning, she
ran away from you laughing, no matter how exasperated you got. Imagine
if she insisted on picking out her own clothes and you let her, rather than
fight about it. Imagine if she could sing the entire theme song to "Golden
Girls," could go down the slide on her own, could pee on the potty, catch
a ball, dance and chase her friends. Imagine when you step off the subway
after work and walk into her daycare room, all the kids turn to look at who
has entered the room, and when she sees you she flashes the most brilliant
smile and comes running with her arms up, saying "Mama! Mama! Mama!"
Imagine if no matter how many times she had a tantrum and demanded
things from you and exhausted you, she ended each night with a snuggle and
a kiss and you breathed in the smell of her curls and felt warm happiness
all over. Imagine if you could never love anything as much as you loved
your first born child, your dream come true, your daughter.
Now imagine it’s 9 months later. Imagine she is lying next to you in your
bed. She can't walk. She can't use her arms or hands. She can't hold her
head up. She can't see the television. She can't tell you she loves you.
She can't hug you. She is lying in the bed sound asleep, but coughing on
her own saliva, which she is starting to choke on because she can barely
swallow. Imagine she was dying and there was nothing you could do to
change it. Imagine if you knew that one day soon you would never get to
see her again. Never see her smile, feel her hand slip into yours, kiss her
warm cheek, feel her sigh into your chest.
That is the simple reality of what we are living with. And it's hard. No matter
how many good things happen to us, no matter how much we believe in a
bright future for ourselves and a time of healing, we are being tortured. No
matter how well or easily we manage to get through the days, to talk with
our friends, to laugh and joke and even fight sometimes, we are broken
inside. It's a very strange way to live. We need to not focus only on what
we are losing, but on all we have gained, but despair creeps in nonetheless.
Chapter 21: Integrating Palliative Care
318
It is never the same in progress…always the same in result. It is the cessation
of the brain signal to breathe that causes the death of each child. That
time can come swiftly as with my daughter Savannah…4 1/2 months from
diagnosis with absolutely no slowing of symptom progression…or it can
move more slowly like in so many kids. I hate it…I hate it!
I think I've said before that despite the large groups of people who surround
us, sometimes grief feels incredibly lonely. One of the realizations I've made
this past week, is that there never seems to be a good time to cry. All these
people come in and out of our house and they bring hugs and kind words
and delicious foods and generosity and beautiful friendships, but no one
ever comes in and just cries. I guess it's human nature to want to avoid
being sad in front of one another, but I feel like crying all of the time and
it's difficult to find the “right time" to do it. You can't do it when you're out
walking on the street…you shouldn't really do it when you have company…
it upsets our daughter if you cry in front of her. The only safe spaces left
are at night when the darkness blankets my room and I fall asleep with hot
tears pooling at my neck, or behind my dad's house on a small swing that
lies hidden from sight.
This is the saddest thing in the world, but no one wants to cry with me. I
understand it on some level, but sometimes it makes me feel as though no
one else is sad, or they are able to push it out of their minds. I am jealous.
I wish I could also ignore the grief, but to me it's palatable in the air we
breathe day in and day out within the white walls of our house.
I wonder if the issue is that we live in such a superficial culture that often
seems uncomfortable with true depths of feelings, in particular grief. I feel
there is a certain amount of intolerance of acute sorrow and intense mental
anguish that makes up the bulk of my life right now. Sorrow is something to
be medicated, as I'm doing right now, or something to be divided into five
recognizable stages that I can read about, label and rate my growth with.
Grief is too complex an emotion to be ignored, pushed away, or forgotten
about. I have been grieving my daughter since June 24th and have learned
that for me to grieve is to let sorrow and tears invade my soul so that it
permeates my pores like a heavy perfume. I am always stunned that no one
else can see and smell the sadness that is so obvious to me.

319
Chapter 22: Journey of Sadness and Hopes
Chapter 22
Journey of Sadness
and Hopes: A Letter to
Parents
Tammy I. Kang, MD, MSCE
Chris Feudtner, MD, PhD, MPH
Dear Reader,
As this book draws to a close, following many pages lled with information
about the diagnosis, prognosis, scientic advances, and emerging treatment
options for diuse intrinsic pontine glioma (DIPG), what can we add that would
be worthwhile to parents? Is there anything we can say that would help in this
journey of sadness and hopes?
After wondering and worrying about these questions, we thought that the best
place to start would be to tell you, from the bottom of our hearts, that we wish
your child did not have this cancer. We wish that you did not have to embark
upon this journey. We wish that we knew with certainty what your questions and
concerns were, that we understood what you might nd potentially helpful. And
we wish that we knew you, so that we could say all of this face-to-face.
We decided that the best way to proceed was to write the nal entry for this
book as though we were writing a letter to distant friends who had asked for our
thoughts and suggestions. us we write with the hope that some of what follows
may be of help to your child, to your family,
to you. At the same time, we know that no
single conversation, no single approach,
can work for everyone; so if our ideas aren’t
working for you, please put this aside and
accept our apologies.
Dr. Kang is the Medical Director
of the Pediatric Advanced Care
Team at Children’s Hospital
of Philadelphia, and Assistant
Professor of Pediatrics at the
Perelman School of Medicine at
the University of Pennsylvania,
Philadelphia, PA.

Chapter 22: Journey of Sadness and Hopes
320
321
Chapter 22: Journey of Sadness and Hopes
e Breadth of Hopes
If we were able to sit down and talk, we might ask a question that we often pose
to parents as a way to start a conversation. Given your child’s situation, what are
you hoping for? ere are many answers to this question, and none of them are
wrong. We encourage parents to relay as many hopes as they have. is takes time.
Some hopes may feel much more important than others. When working with this
question, at this point, the goal is to get all the hopes to surface and to set them
out in front of us, as though spread upon a table, so that they can be discussed.
Indeed, some of our patients want to do this. Once, a cheerful, bright-eyed
11-year-old girl, meeting her oncologist for the rst time said, “Before you start
asking me a lot of questions, you should know a few things. I know I have a
brain tumor called an intra-pontine glioma. I know most people can’t be cured
of this and that I have had a lot of problems because of it, more than most. I was
in the hospital for a long time and didn’t like it much. I already had radiation
and am excited to move here because it will be easier here for my mom.” As if
this wasn’t already remarkable enough, she nished by saying, “I hope for lots of
things, some of them maybe I can have, and some I can’t. Even though I know my
cancer probably won’t go away, there are still things I want, like I would really like
to be able to get rid of this trach and g-tube before I die. I want to go to school
and, no oense, I’m sure you’re really nice and everything, but I would rather
not see you too much.” Remarkable. In one conversation this patient illustrated,
and illuminated what we have learned over decades—that inherent within us,
no matter how dicult the circumstances, we harbor hope. Not a singular hope,
but many hopes.
Hope for the best
One of these hopes can always be—and, in our conversations with parents, often
is—to hope for the very best. ese hopes take many forms, and are often the
rst thing that parents mention when asked about their hopes. Some of these
hopes are as large as life, or even larger.
I hope that the cancer goes away. I hope
that the treatment cures this awful disease.
I hope that this is all a bad dream. I hope
for a miracle. Parents also have other hopes
which, in some situations, may seem quite
small in comparison; but to those who
hope such hopes, they brim with life. I
hope to go home. I hope my child gets this
tracheostomy and gastrostomy tube out. I hope to celebrate my child’s birthday.
I hope to see my child with those whom I love and who love my child. Hoping
for something that may not happen does not mean you “don’t get it” or have
unrealistic expectations. For many parents, these hopes are one clear way they
show their love and devotion for their children. And while we cannot say for sure
for you, for many parents, there are other hopes as well.
Hope against the worst
Hopes can also be shields of sorts against the potential worst. We cannot begin
to imagine the emotions that have entered your life. Parents have shared with
us their intense, painful feelings of fear, anger, desperation, and sadness. Hopes
swirl aloft in these wild winds. ey do not tame the feelings, but they can help
to ride them out. Some hopes will be that certain events do not happen. Other
hopes will be that, if those events do happen, plans will be in place to assure that
the child is well cared for, as protected as possible, comfortable and not afraid.
Modifying the old adage, hope for the best and plan for the worst so that you
can hope that the true worst does not happen. Although dicult to discuss these
fears, mentioning them to health care providers and asking, “What would we do
if this happened?” can start the process of making these plans tangible, forging a
clearer pathway toward the hope that the worst will be prevented. Although the
hope that arises to counter these fears may seem grim, this type of hope is full of
deep commitment and resolve.
Everyday hopes
When the diagnosis of DIPG enters the lives of many families, for a time life
may seem to stop, to be put on hold, consumed by the chaos and emotions
surrounding the diagnosis and coordinating and coping with the early stages of
complex medical care. How can life resume? ere is no single right answer, and
every potential answer is dicult; yet one recurring answer we hear from parents
is this: one small everyday hope at a time. Start by hoping to simply take a deep
breath and let it out. en hope to get outside and see the sun straight overhead
or the rst stars at dusk. Parents have told us that even seemingly insignicant
things like running a short errand without worrying about not being at their
child’s side, or being able to attend to another child without feelings of guilt, is
something to hope for in a day. Name these hopes to yourself and to others, and
then work toward them. One small hope at a time can make a large dierence.
Ask for help if needed and accept the help that is oered.
Dr. Feudtner is an Attending
Physician and Director of
Research of the Pediatric
Advanced Care Team at
the Children’s Hospital of
Philadelphia, and Associate
Professor of Pediatrics at the
Perelman School of Medicine at
the University of Pennsylvania,
Philadelphia, PA.
Chapter 22: Journey of Sadness and Hopes
322
323
Chapter 22: Journey of Sadness and Hopes
Hopes to be a good parent
Many parents we have spoken to wonder—and worry—about what they need
to do in order to be, in their own view, a good parent for their child with serious
illness. is is usually not a topic of conversation that comes up spontaneously,
arising only when we ask whether parents think about this, and then they often
say words to the eect of “yes…all the time.” Cast upon a journey that no parent
ever anticipates, and none are prepared for, parents often have told no one about
these furtive thoughts, which arise from confronting the uncharted enormity of
this disease and the inevitable doubts and confusions. Our hope when broaching
this topic is simply to be a companion and help to dispel some of the loneliness
that accompanies these private worries. What do I need to do? Am I doing the
right thing? Am I doing enough? Again, there are no right or wrong answers.
We nd that parents usually can name a few things that they feel that they must
do for their child, and hope fervently that they can do these things. Our job, as
we see it, is to gain an appreciation of what these things are, help the parents to
achieve what they are hoping to do, and oer feedback and reassurance that they
are indeed doing them.
Hopes for health care providers
“Given your child’s situation, what are you hoping for?” Sometime during the
days or week after receiving the news that their child has a DIPG, when the
initial shock of the news has started to lift, this question should be routinely
posed to parents. Yet typically, medical providers don’t ask. Is it because they
are sure they already know the answer? Because they cannot bring themselves to
imagine with this family what hope might look like in even the most dicult of
circumstances? Or is it because they are afraid of not being able to aid in making
those hopes a reality?
Quite often physicians assume—or, more blatantly, assert—that parents have
only the capacity for one hope: hope for cure, the hope for a miracle. In this
regard, physicians and other health care providers are not alone. Studies of the
social construct of hope as portrayed by print media suggest that the public
dialogue in the newspapers and magazines around hope for patients with advanced
cancer conveys the message that only one legitimate hope exists for persons with
cancer—hope for a cure.
What are the origins of this notion that, when given a diagnosis of advanced
cancer, hope becomes a singular entity? Perhaps this concept is just a self-reassuring
shared myth, consoling all of us, who do not have to confront cancer personally,
that one need only hope for cure. Perhaps this notion is a way to push aside
the health care provider’s own feelings of failing their patients and families. Or
perhaps this is an optimistic—and thus eective—way to market and advertise
medical innovations.
Whatever the origins, this notion is a conceptual straightjacket, causing us to
underestimate a parent’s ability to harbor, frame, and hold hope while still being
grounded in reality. Certainly parents have voiced that what is important to
them is honest communication with their child’s medical providers about their
child’s medical issues, regardless of prognosis. Health care providers may need to
be encouraged that one of the most valuable things they can give to patients and
parents is permission to hope—and that they themselves can hope to participate
in the care of a patient and family, oering to help in ways that extend beyond
the physical or biological treatment of disease, all in the pursuit of hopes.
Completing a Circle of Hopes
Not all of the aspects of hope outlined in this chapter may come readily to you
or your child or your family. And some aspects may not be what you need. What
is right, hope-wise, is what is right for you and your child. In the profusion of
what you hope for, your hopes are intertwined with your values, your loved ones
and your goals. Take a look, from time to time, at what your hopes have become,
building your hopes until you can envision your child, your family, and yourself
surrounded by a protective circle of hopes, 360 degrees of commitment and
compassion, capable of moving forward with purpose.
We started by asking whether there was anything that we could do that would be
helpful, and here at the end we still worry that our words may not have suced
to bring clarity of thought or comfort of feeling to you. Yet we, too, live in hope,
as we all must, and hope that you have found something of value in this letter,
something within you or around you that can help carry you, your family, and
your child forward with hope.
Chapter 22: Journey of Sadness and Hopes
324
325
Chapter 22: Journey of Sadness and Hopes
Parent Perspectives
Julian was diagnosed with an atypical brainstem glioma because of the
way his tumor was growing up and out of his brainstem. We clung to the
hope that this tumor would prove “atypical” in its behavior and that he
could be an exception to the grim statistics.
Another brain tumor parent asked me, “How do you walk the line between
accepting the reality of this and staying hopeful?” It was the internal
struggle we faced every day. Walking the tight rope between fear and
optimism, we found balance by taking it practically one hour at a time,
savoring the small, beautiful moments and committing to the task of keeping
him feeling safe and loved.
Early on, a friend and pastor made me tell him the prognosis. He said,
“You need to be honest with me before I can help you.” It was the first time
I said it aloud, “Typically patients have less than a year.”
His advice that followed stuck with me throughout our 7-month battle.
He said, “You need to prepare your heart, but make no plans to lose.”
Encouraging us to stay hopeful, he continued, “You will not start planning
the funeral. You find him the best care you can, and you will not stop
searching for a breakthrough.”
Keep your head where your feet are was my motto. You can spend your
time searching the world over for a wonder cure or treatment, but if you
use your time wisely you will quickly see what is important. Assign that
role to someone else. Keep in mind your priorities and don’t lose sight of
quality over quantity. Don’t ever lose hope but please put reality in the
driver’s seat so that you don’t miss out on one single happy moment with
your child. Ask for your miracle but some part of you must come to terms
with the “what if you don’t get it” part. Don’t think you are giving up if you
decide to end treatment or follow a different path. You are never going to
give up. You are always going to do what you think is best for your child.
It was at the end of radiation that we did another good thing. We were
asked, and agreed, to meet with the pediatric palliative care nurse, before
going home. I can say that I was in complete dread of meeting Bryce’s
palliative care nurse, because she was going to tell us all the terrible
things that we couldn’t and didn’t want to even imagine yet, including
how and when Bryce would die. I wasn’t ready for it, didn’t want to hear
it. The day that my husband and I met with her I was physically sick. We
walked in, and the first thing that she asked us was to tell her about Bryce,
which threw me off kilter. She didn’t want to know about his symptoms like
every other person we had encountered. She wanted to know about him as
a person, and as a child. She wanted to know what his interests were, and
about our family. After being submerged in the cancer world and on “a
mission” to treat him, this was somewhat shocking, and even refreshing.
She then looked at us and asked if we knew what palliative care was all
about. We said yes, it was about dying. She said, yes, that was part of
it, but it was also about so much more. She looked at us and told us that
palliative care was about choosing to LIVE, for as long as possible, and
in the best manner possible with the time that is left. It was about making
each day a choice to live, not about waiting to die. And at that moment
we made a choice. We decided that we would do just that. We would go
home and LIVE Bryce’s days with him, and make those days everything
that they could be—and it became our new mission. Our hope changed.
We knew there was no cure, but we also knew that our hope became about
making whatever days we had left with our son be good ones. Those days
would have to be so good that for him, and for us, and for his sister there
would be no regrets.
Once we were home, we contacted hospice right away. We were trying to
find a connection at home for Bryce and for our family that was local. We
all started to see the social work team there. It was one of the good things
that we did. Bryce met with the social worker every couple of weeks. She
suggested that they work on a scrapbook together, one page every session,
about Bryce’s life. So we would get fancy papers and pictures ready for
the theme of the week, and during their sessions, they would talk—alone.
It was nice for Bryce to have someone to talk to who was not a family
member, and to have someone to tell how he really felt about what was
happening, without being afraid of hurting anyone at home. They worked
on the scrapbook for almost 6 months.
Over that summer and into the fall many wonderful things happened.
Chapter 22: Journey of Sadness and Hopes
326
327
Chapter 22: Journey of Sadness and Hopes
Bryce got his “Wish” for a camper from Make-a-Wish Foundation. We
camped and traveled. He spent time with his friends, and he did his
best to be a regular kid. He continued to have double vision, so some
activities were no longer possible, but he was determined and found ways
to compensate. His balance generally improved, so he still rode his bike.
As dirt-biking was too fast, much to the dismay of his doctors, we got
him a four wheeler, which he was allowed to ride on flat fields with his
friends. This was probably his most prized possession that summer. For
the most part it was a good summer, when he felt good.
He never really wanted to discuss his illness except on the days when he
wasn’t feeling well. He even started high school. He attended two classes in
the morning. He went to the gym with special permission in the afternoon
with his dad, and we got him a personal trainer. This really helped to
keep his strength and balance up, too. He continued to hang around the
arena, skated when he felt up to it, and even coached his own team with
his father. He always amazed us with how hard he fought to be a regular
kid every single day. He wouldn’t accept any less from the rest of us either.
Not only were we on a mission to help Bryce no matter where this journey
took our family, but we were also dealing with the reality of losing a
child. And you do what you have to do to help your child, no matter
what—because that’s what he expected of us. There were many days when
I would just want to crawl into bed and never get up again. We were
already grieving and he was still here. But he would say, “Mom, go take
a shower, you look like crap.” And I remember crying one day all the way
home from the hospital, and cuddling with him when we got home that
night. He was accepting of me being sad, but then, he suddenly looked at
me, and said “Mom, you can cry today, but you can’t cry tomorrow.” I
asked why not. He said, “Because I just need you to be mom every day.”
And that was when I decided that no matter what happened, if my son
was going to have to do this, that I would be present every step of the
way. What choice was there? He needed us to be.
It was at this point that we decided that we needed to fill in a booklet
that was given to us about what Bryce wanted for end-of-life care. As
Bryce’s progression developed, his speech became quite affected, so we
were glad to have this booklet full of answers to help us to make sure that
his wishes were being met. It also helped for planning his celebration of
life thereafter.
As Bryce lost his gross motor ability to hold up his head and to sit, we
found the right wheelchair for him so that we could still do things with
him. He wanted to go for a walk around the block one day. We had to
wait until all visitors left, because they thought it was a terrible idea
taking him out. It was unsafe, he would get sick, they argued. But that
was what he wanted. These adventures took 2 to 3 hours from start to
finish. He wanted his snowsuit on because it had snowed. So we did that.
We went for a walk, and he wanted us to lay him in the snow. So we did.
He wanted to lie in bed with his hockey skates on, so he did. He wanted
to hold his skateboard in bed, so he did. He wanted a remote control car,
so we searched the earth and found it in Australia. He got it with a lot of
help. He wanted to go to Walmart for an F150 dinky car. So we did. One
of the biggest adventures was that Bryce wanted to go and watch his sister
play hockey. It was her first hockey tournament. The arena where she was
playing did not “support” a viewing area for individuals in wheelchairs,
so, with the help of Earth Angels, 6 police officers came out to lift Bryce
and his wheelchair up a flight of 25 stairs to the viewing area. He watched
his sister play, and at the end of the game he said, “You forgot to tuck in
one side of your jersey, Bailey.” Those officers came back an hour later
to lift him back down. Another day former NHL player Bobby Orr showed
up at our house to visit.
I have to say that as a parent, and not a health care professional, it was
the scariest thing that we will ever experience, but we could only have
survived it because of the communication and support that we received
from the healthcare team. You see, for the entire 361 days that Bryce
fought this cancer, we were supported. We were able to talk to people who
could help us and explain things to us, so that we could in turn explain it
to Bryce and his sister, and ultimately live through what was happening.
Everyone was only a phone call away, or a visit away. Once in Care at
Home, Bryce’s care team continued to support us by calling us regularly
in the evenings. This meant so much to us, because this would be when
we felt that much more separated from medical staff and when truthfully
after dealing all day, we were most vulnerable and scared of what was
to come. What if it happened at night?
I can tell you that now, 20 months later, I still feel that we did what
was right for our Bryce and for our family. All of the decisions and
conversations that we had were right for us. We have no regrets that we
didn’t get to talk about things, or that we didn’t get to spend time doing
Chapter 22: Journey of Sadness and Hopes
328
329
Chapter 22: Journey of Sadness and Hopes
the things that Bryce chose to do, or that he didn’t LIVE every single day
that he was given. That is not to say that we do not miss our boy every
second of every day, or that our life will ever be the same without him.
We said what we needed to say, and did what he needed us to do. In the
meantime, we LIVE as he showed us how to—with hope for a cure, much
courage to get through our days, strength in making good decisions, and
with the peace of knowing that we will one day be together again.
As I look back on this journey, my advice to parents that may have a
newly diagnosed child with DIPG is to not give up. There are survivors
of this—not many. It is crucial, in my opinion, to send out your child’s
scans to doctors that know this tumor. Join the DIPG yahoo group online
for a list of exceptional doctors with expertise in DIPG, for other families
that have gone through this and are in the battle, for the most up-to-date
treatment options from around the world, for clinical trial information, for
alternatives that others have tried, and for understanding. Please know
there are people that will help you and will listen and will understand.
Finally know that as the parent you will make the right decisions for your
child in this very difficult journey.
Things were as normal as they could be with all the appointments and the
anxiety of knowing that this reprieve was probably going to be a short-
lived gift. Although we held out hope that Miguel would be the one to
beat this, we were realistic. We were able to enjoy him fully for 18 months
from diagnosis. It wasn't easy, especially when progression occurred
almost one year from diagnosis, just like the doctors said it would. While
he was able to compensate for many of the symptoms prior to diagnosis,
after progression, things changed one by one: loss of mobility, difficulty
swallowing, loss of speech, and the loss of fine motor skills to name a few.
Although the deterioration was difficult to watch and endure, Miguel's
short life has made a profound impact in many lives. Miguel is a precious
yet rare gift and it is an honor to be his grandma—always.
“Honey, I am scared Johnny is going to die too. Right now, I don’t know
if he will or not, but I do know he isn’t going to die today. So each day I
have with him, I am going to love him and make it as special as possible.
Does that sound like a good plan?” At this point, we were both crying.
Through our tears, a small smile came to her face and we headed back
up to the room to check on the one boy we couldn’t stop thinking about.
We had been at St. Jude for about three weeks. We had a routine of
radiation, chemotherapy and clinics. We were beginning to accept that
Johnny had cancer, and although there was no cure, we were given more
time with him, and we all wanted more time. One afternoon, we were in
the truck driving back to the Ronald McDonald House. Johnny’s stomach
was upset and he was lying down in the back. Johnny speaks up from the
back, “Mom, am I going to die?” I felt the stabbing dagger of pain once
again. My son knew he was going to die and no one needed to tell him,
but he needed to talk.
Since I had already talked with my daughter, I knew how I would respond.
I went on to ask more questions from him and then closed the conversation
by telling him his feelings were normal and good, his dad and I felt the
same way, there were some things we can’t control and don’t have the
answers to, how much we love him and how special we will make the time
we do have with him. We were taking life one day at a time. We had no
guarantees about tomorrow, but today we were living it for all it was worth.
The questions from my other children continued, “It’s not fair. Why did
he get sick? What did we do wrong? We are good people. I mean we do
some bad things but not really bad. Why is God punishing us?” I didn’t
know what to say. Having questions about faith and spiritual matters is
normal. The intensity of this pain is going to bring to the surface questions
that most people can go a lifetime without addressing. One option is to
ignore or avoid my child’s questions. A second option is to seek out the
answers and wrestle with the questions. Option one gives immediate
relief but the questions will resurface throughout their life because they
go unresolved and unanswered. Option two is a more difficult path, but
when the wrestling is over and the tension is resolved my other children
will be able to move through their grief in a healthy pattern.
So, this was my reply. “I don’t know why God is allowing this to happen
to us. This is what I do know. I know all the stories of people helping us.
I know dad’s friends from his childhood live in Memphis and have a house
so you kids can stay here with us. Most families have to live apart from
each other all this time. I know that grandpa and grandma have the time
to live here with you, so that dad and I can take care of Johnny. I know
Chapter 22: Journey of Sadness and Hopes
330
331
Chapter 22: Journey of Sadness and Hopes
dad’s work is letting him stay here the whole time Johnny is at St. Jude
and still paying him so we don’t lose our house. I know our neighbors are
mowing our grass and taking care of the dog. I know people are sending
you kids gifts and cards. I know that every time we turn around someone
is doing something special for us, and a lot of those people don’t even
know us because we just moved to Arkansas three months ago. I know
these things mean something."
I could not offer many answers to my son that day. I still cannot offer
many answers. But my answer offered hope to him. Hope that there are
good things in the face of tragedy. Living one day at a time, hope was
enough to calm my child’s heart.
In the days that followed his diagnosis we would tell Liam he had cancer.
We felt it was incredibly important to tell him the truth, to use that word.
As hard as it was, we wanted him to know he could always trust us to be
honest with him going forward.
We called family and close friends. We had family bring our other three
children to the hospital. We told our kids that Liam had brain cancer and
that they could not take it out but that we were going to do everything we
could to make it smaller. Liam's neuro-oncologist allowed the kids to visit
Liam very briefly. Liam's identical twin brother asked his doctor what
would happen if the medicine didn't work. The doctor reassured him that
they had lots of things they could try to help Liam feel his best. We felt
grateful for this small offering of hope.
I would have liked to have a group of individuals to talk to about our
daughter's specific tumor type during her treatment. We were following
another DIPG child’s story and were very inspired at how long he was able
to live after treatment. We never gave up hope and we always believed that
something would come up to save our daughter. We never really thought
she would really die from this even though that's what they told us.
Things are changing for our kids, for your child. There are viable
alternatives to current steroids and new therapies on the horizon. I know
that there is always hope.
We have found it incredibly helpful to hear from other parents that have
lost a child. Their advice is always so heartfelt and honest. The only
advice we can come up with at the moment is to listen to your own heart
and make decisions based on your knowledge of your child, individual
circumstances and personal convictions. Grieving is such a personal
journey, and the patience you need to have with yourself is infinite. No
one's path with DIPG is the same, and the only thing you can strive for is
that you don't regret any of the decisions you've made. We think the best
way to do this is to trust your instincts and be at peace with the journey
as much as possible.
Stella is many things. As was evident early, she is a force to be reckoned
with. She is inquisitive. She is intelligent. She is hilarious. More than
anything else, Stella is her aptly given middle name: Stella is “Joy.” Stella
proves that cancer can't take away everything...her smile is our lifeline!
Aimee asked me last night if I thought we would ever be happy again.
It's an interesting question. It's not that we haven't felt happiness these
last 8 1/2 months; it's just that all happiness is tinged with a pervasive
sadness as well. I had to think for a while before answering, because the
truth is I have no idea if I will ever feel true happiness again. I think
we will definitely have many moments in the future of being happy. But
feeling happy? I think they are two different things. My best guess is
that each joyful moment will come with a small shadow of wishing that
our daughter was there to experience it as well. Every family vacation
and holiday, every accomplishment in our lives, every celebration (big
or small) that she should have been there for. I imagine that eventually
the shadow around your heart just becomes who you are and happiness
and sadness cease to exist as separate entities and your new norm is to
just accept that happiness and sadness are not mutually exclusive, but
intertwined in one another—bitter and sweet.
What is keeping us moving forward right now, even when our hearts are
completely broken, is watching how our daughter has chosen to live her
short life. How she treats each day as a new adventure, pushes herself
both physically and mentally to ensure that she accomplishes what she
wants on that particular day. Sometimes it's something big—painting
Chapter 22: Journey of Sadness and Hopes
332
with her mouth and visiting the pigs at the farm. And sometimes it's just
being able to mouth the words "ice cream," and then napping most of the
day. But she is always true to herself, and even though things are hard
for her, she ignores the barriers of DIPG and chooses to forge her own
path. Most importantly, she believes that when life gives you a hundred
reasons to cry, you need to find a thousand reasons to smile…And in my
own smiles, I have become familiar with the bittersweet taste of getting
to parent my precious daughter—the best experience in the world, but
like a spring day that is much, much, too short.
333
Appendix A: Medications Form
Appendix A
Medications Form

Appendix A: Medications Form
334
Appendix A: Medications
Name of
Medication and Strength
Instructions/Dose
(Include all
details, dose, and times)
Purpose Prescribing
Doctor
Ex: Tylenol liquid
(acetaminophen)
160 mg/5 ml
Takes 3 ml every 4 hours by mouth
as needed
For fever or pain Dr. Smith
It is strongly recommended that you take any medication that your child is receiving at home to each doctor or hospital visit.
e medications will be reviewed and any changes can be made at that time.
335
Appendix B: Glossary of Terms
Appendix B
Glossary of Terms
e following terms were referenced by the authors throughout the book. ey
are listed here to provide additional information to assist with understanding.
Accessible: Tumors that can be approached using a surgical procedure.
Active Immunization: is is the kind of vaccine we are most used to. e
MMR, inuenza, DPT, and Polio vaccines all depend on active immunization by
presenting an antigen to the immune system and inducing an immune response
and long-lasting central memory.
Adaptive Immunization: is comes into play when the innate immune system
is evaded and an invader (a pathogen or cancer cell) gains a foothold. e adaptive
response recognizes these invaders and enables the immune system to mount
a stronger response each time they are encountered. is response involves all
branches of the immune system, both B cells and T cells, working together in
balance. e adaptive response can be trained through dendritic cell vaccine
strategies.
Adjuvant Chemotherapy: Administering chemotherapy after the primary tumor
has been treated by some other method, for example after radiation.
Adoptive Immunization: e transfer of mature circulating lymphocytes to
treat certain diseases.
Anaplastic Astrocytoma: A synonym used interchangeably for WHO grade 3
astrocytoma (glioma).
Angiogenesis Inhibitor: An agent that inhibits the growth of new blood vessels.
Apraxia: Inability to perform activities such as making gestures, speaking in spite
of the person’s willingness to do so; inability of the brain to correctly communicate
instructions to the body.
Astrocyte: One of two types of glial cells in the central nervous system that help
support the neural cells. e other type of glial cell is called an oligodendrocyte.
Appendix B: Glossary of Terms
336
337
Appendix B: Glossary of Terms
Astrocytoma: A tumor developed from the glial type of cell called an astrocyte.
ese tumors are oftentimes further described by location or appearance under
the microscope. e microscopy appearance can be into one of 4 grades using
the World Health Organization classication.
Ataxia: Uncoordinated and clumsy appearing walk that is associated with balance
issues.
B Lymphocytes: Cells in the immune system responsible for the humoral immune
response. at is the production of antibodies that attack foreign antigens (like
bacteria) or tumor associated antigens.
Blood-Brain Barrier (BBB): Protective barrier separating circulating blood from
the brain's extracellular uid thereby preventing substances in the blood from
entering into the brain. e BBB is created through tight junctions around the
capillaries of the linings of the blood vessels of the brain.
Brainstem Glioma: e broadest term to describe all histologic grades of glial
tumors that are located in any part of the brainstem (pons, medulla, tectum
and cervicomedullary junction). Brainstem gliomas can always be classied
more specically by particular location (e.g. pontine glioma, tectal glioma,
cervicomedullary glioma) and by certain descriptive terms (e.g. diuse, focal,
intrinsic, exophytic and extrinsic).
Biopsy: Surgical procedure to remove a sample of tumor tissue to establish
diagnosis. Tissue can be further utilized to determine specic molecular analysis
for research purposes as well as potentially used to develop personalized treatment
plans.
Cerebellar Ataxia: Loss of muscle coordination brought on by a lesion in the
cerebellum.
Cerebral Spinal Fluid (CSF): Clear, colorless uid that circulates around and
inside the brain and spinal cord.
Contrast Enhancement: Use of an agent administered to a patient prior to an
MRI scan to increase the visibility between the tumor and surrounding tissue.
Computerized Tomography (CT) Scan: A medical imaging procedure using
x-ray technology from a series of dierent x-ray angles, which are then processed
through computer technology to create cross-sectional images of bones and soft
tissue including the brain; also referred to as CAT scan.
Cranial Nerves: Twelve pairs of nerves originating in the brain, and often included
in the designation of central nervous system (brain, spinal cord, and cranial nerves.)
Cytoarchitecture: e typical arrangement of cells within a particular tissue or
organ.
Cytotoxic: Toxic to cells. Any agent or process that kills cells.
Cytotoxic T Cells: T lymphocyte cells that directly induce the death of tumor
cells or virus-infected cells; also known as killer T cells.
Daughter Cell: Cell(s) that result from cell division. Daughter cells are genetically
identical to the originating parent cell.
Dendritic Cell: ese cells are unique to mammals and function to capture
foreign invaders (antigens) and present them to the immune system. Activated
dendritic cells from the nose, lungs, or skin migrate to the lymph nodes to tell
B and T cells what to do.
De Novo: New—for example a de novo mutation is a new genetic mutation.
Diuse: An adjective that can be used to describe an inltrative nature of a
tumor as opposed to focal tumors which are more conned or circumscribed.
Diuse tumors are usually a higher histological grade (3 or 4) but can be low
grade. Except in the cases of leptomeningeal spread or gliomatosis cerebri, diuse
is also intrinsic. ese tumors cannot be removed as they are like "sand in grass"
or "pepper in Jello" i.e. being too dicult to remove the tumor without severely
disturbing the normal tissue of the pons.
Diuse Brainstem Glioma: is is an inltrative tumor of glial origin located
anywhere in the brainstem. Approximately 80% of these tumors will be in the
pons. us most, but not all, diuse brainstem gliomas can be more specically
called diuse pontine gliomas or diuse intrinsic pontine gliomas.
Diuse Pontine Glioma: A glioma (usually an astrocytoma) located in the pons
which intermingles and inltrates normal pontine tissue. Synonyms include
diuse intrinsic pontine glioma or diuse inltrative pontine glioma.
Diuse Intrinsic Pontine Glioma (DIPG): A glioma (usually an astrocytoma)
located in the pons which intermingles and inltrates normal pontine tissue.
Synonyms include: diuse pontine glioma or diuse inltrative pontine glioma.
Diplopia: Double vision
Dorsal Exophytic Pontine Glioma: Tumor that grows from subependymal glial
tissue out into the 4th ventricle; typically presents with hydrocephalus.
Dysarthria: Speech disorder resulting from defects in the central or peripheral
Appendix B: Glossary of Terms
338
339
Appendix B: Glossary of Terms
motor nerves leading to an impairment of neural transmission to the muscles
involved in speech. Can include impairment to all processes involved in the
production of speech including respiration, phonation, and articulation.
Dysphasia: Loss or impairment of the ability to speak, read, or write; understand
or interpret speech or written language.
Edema: Excess bodily uid leading to swelling.
Embryo’s Yolk Sac Endoderm: e germ layer that lines the yolk sac.
Endoscopic ird Ventriculostomy (ETV): Surgical procedure in which an
opening is created in the oor of the third ventricle using an endoscope through
a burr hole. is allows for the cerebrospinal uid to ow directly into the basal
cistern, thereby used as a means to treat obstructive hydrocephalus.
Extrinsic: An adjective to describe a tumor that is located on the outside.
Exophytic: An adjective to describe a tumor that is growing out of the brainstem—
like the top part of an iceberg sticking out of the water.
Focal: An adjective used to describe a tumor that is well dened and does not
seem to intertwine with normal tissue. Focal brainstem gliomas are usually
histologically grade 1 (also called pilocytic astrocytoma).
Glial Cell: One of the supportive cells in the central nervous system. ese can
be either astrocytes or oligodendrocytes.
Glioma: A tumor arising from glial cells, either astrocytes or oligodendrocytes.
If one can dierentiate the cell line from which the tumor derived from, then it
may be called by a more specic name, i.e. an astrocytoma, oligodendroglioma or
a mixed tumor. Dierent adjectives can be applied to the term glioma to convey
a more descriptive, specic understanding of the tumor.
Glioblastoma Multiforme (GBM): A synonym used interchangeably for WHO
grade 4 astrocytoma (glioma).
Helper T Cells: T lymphocyte cells that help the immune system recognize what
to attack.
Hemiparesis: Muscle weakness on one side of the body.
Hirsutism: Abnormal hair growth on face and body.
Histologic: A term referring to the classication of tissue based on microscopic
examination. With gliomas, there are 4 grades based on the WHO classication.
Hydrocephalus: Excess uid in the brain resulting from a blockage of the CSF
pathways.
Hyperphagia: Abnormal increased appetite for food.
Hypoxic Cells: Cells that are deprived of oxygen.
Hypoxic Cell Sensitizers: Compounds that selectively sensitize hypoxic tumor
cells to the eects of radiation.
Immunohistochemical: Pertains to an assay used in research analysis that shows
specic antigens in tissues through the use of uorescent markers.
Inltrative: An adjective used to describe a tumor that is intermixed with normal
tissue. A synonym for diuse.
Intrathecal: An area sometimes used to administer drugs, which is located in
the space under the arachnoid membrane that covers the brain and spinal cord.
Intratumoral: An area within a tumor.
Intraventricular: An area located within a ventricle of the brain.
Intrinsic: An adjective used to describe that a tumor is located on the inside.
In Vitro: A preclinical study or experiment done within a test tube or laboratory
dish.
In Vivo: A study, medical test, or procedure that is done on a living organism,
such as a laboratory animal or human.
Lumbar Puncture (LP): Insertion of a needle into the subarachnoid space of
the spine to either administer drugs or to withdraw a sample of CSF for biopsy;
also referred to as spinal tap.
mAB: Monoclonal antibody.
Magnetic Resonance Imaging (MRI) Scan: Medical imaging technique that uses
magnetic eld and radio waves to generate computer imaging data; produces high
contrast images of the soft tissue of the body with useful application to the brain.
Malignant: Another term meaning cancerous.
Memory B Cells: e cells that most childhood immunizations depend on.
Memory cells are created from activated B cells the rst time an antigen is
encountered (like a tetanus vaccine). When one encounters the same antigen again
(like stepping on a rusty nail), even years later, the memory B lymphocyte cells
Appendix B: Glossary of Terms
340
341
Appendix B: Glossary of Terms
will respond quickly to create an immune response before things get out of hand.
Memory T Cells: Experienced lymphocyte T cells that have previously
encountered virally infected cells or tumor cells. ey are more eective than
naïve T cells are when encountering an immune target for the second time, i.e.
they hit harder and faster. Memory T cells are divided into central memory and
eector memory subtypes.
Mitotic Cycle: e transferring of the parent cell genome through cell division
into two identical daughter cells.
Necrosis: e death of living cells or tissue(s) due to disease, injury, loss of blood
supply, radiation or chemical agents.
Neoadjuvant: Induction therapy that is given to a patient prior to the main
treatment; can include chemotherapy, radiotherapy, hormone therapy. e goal
is to reduce the size of the tumor prior to the radical therapy.
Neoadjuvant Chemotherapy: Administration of chemotherapy in order to
decrease the tumor burden prior to treatment by other modalities such as radiation.
Neurospheres: A free-oating (non-adherent) in vitro spherical cluster of neural
stem cells.
Neurotoxicity: A damaging eect on the nerves or nervous tissue.
Passive Immunization: Fetuses acquire antibody from the mother via the placenta
of breast milk and are more able to cope with specic infections during the rst
weeks of life. Articial passive immunity can be used to treat transplant rejection,
rabies or tetanus.
Peritumoral: Area around the margins of a tumor.
Peritumoral Edema: Swelling around a tumor.
Pilocytic Astrocytoma: e lowest histologic grade of tumor. When in the
brainstem these tumors tend to be more conned and less inltrative. ey
possibly could be operable. ese are WHO grade 1 astrocytomas.
Plasma B Cells: Large B lymphocyte cells that have been exposed to a specic
immune target and make lots of antibodies against it.
Pons: A specic area of the brainstem located below the midbrain and above the
medulla which is connected to the cerebellum through the cerebellar peduncles.
Pontine: An adjective used to describe the specic location as being in the pons.
Pontine Glioma: A tumor of glial origin which is located in the pons.
Progenitor Cell: Cell that is an early oshoot of a stem cell but one that is more
dierentiated than a stem cell.
Radiation Necrosis: e death of living cells or tissue(s) caused by radiation.
Radioresistant: Tumors that do not respond well to conventional radiation
therapy.
Radiosensitive: Tumors that do respond to conventional radiation therapy.
Ras Protein: A protein involved in signal transmission within cells which typically
promotes normal cell division. Abnormal ras, caused by gene mutation(s) results
in increased cell division leading to cell proliferation.
Resect: Surgically remove.
Spiral CT Scan: CT scan technology using a helical/360 degree capture of the
x-ray image which results in increased resolution, also referred to as a helical CT.
Stem Cell: Master cell that is undierentiated within the human body, capable
of growing into any one of more than 200 cell types, allowing them to replace
defective or lost cells/tissues in patients with disease or defects.
Stereotactic: Surgery or radiation therapy that is directed by 3D scanning device
to enhance procedure accuracy.
Sublethal Radiation Damage: Radiation that damages but does not kill the
tumor cell.
Suppressor T Cells: T lymphocyte cells that maintain immune tolerance so we
don’t attack ourselves or overly respond to everything we come in contact with;
also known as Regulatory T Cells (Treg).
Teratogen: Drug that can disturb the development of an embryo or fetus.
T Lymphocytes: Cells within the immune system which are responsible for the
cell-mediated immune response.
Tumor Necrosis: Death of tumor tissue.
Tumor Vasculature: Arrangement of blood vessels within tumors; vascular-
targeted therapies are being studied to destroy the blood supply to cancer cells
within tumors.
Ventricle: A small cavity located within the brain.
Appendix B: Glossary of Terms
342
WHO Grading of Glial Tumors: e World Health Organization has criteria in
categorizing glial tumors by the way they appear under the microscope. ose that
appear closer to normal cells are lower grade (1 or 2) and are generally considered
less aggressive. ose that look more abnormal are higher grades (3 or 4) and are
typically considered more aggressive.
Grade 1-pilocytic astrocytoma
Grade 2- brillary astrocytoma
Grade 3- Anaplastic Astrocytoma
Grade 4- Glioblastoma Multiforme
e number grade and the corresponding terms are considered synonymous and
used interchangeably.
343
Appendix C: Resources
Appendix C
Resources
Many resources exist for families of children with diuse intrinsic pontine
glioma. is appendix contains a sampling of some especially helpful books,
organizations, videotapes, and websites.
Books
General
Keene, Nancy. Chemo, Craziness, and Comfort: My Book About Childhood Cancer. American
Childhood Cancer Organization, 2002. Provides clear explanations and practical
advice for children ages 6 to 12 with cancer. Warm and funny illustrations, by Trevor
Romain, help the child (and parents) make sense of cancer and its treatment. Free
to children with cancer.
Dodd, Michael. Oliver's Story, For 'Sibs' of Kids with Cancer. American Childhood Cancer
Organization, 2004. A practical book written to celebrate the ways in which siblings
of children with cancer can help during this time of family crisis. Free to families of
children with cancer. Available in English and Spanish.
Homan, Ruth I. Along the Way. American Childhood Cancer Organization, 2010. is coil-
bound journal provides a place for contact information for doctors, school and other
caregivers essential to caring for the child with cancer. It also includes information
about clinical trials, informed consent, medical terminology, blood counts as well as
forms to log the child's temperature and out-of-pocket expenditures. An extensive
journal section is also included. Free to families of children with cancer.
Homan, Ruth I. Cozy Cares Journal. American Childhood Cancer Organization, 2010.
is 122 page journal includes illustrations by Trevor Romain. e drawings of Cozy
the 'Port-a-Cat' include hand gestures such as 'high ve,' 'ok,' and 'thumbs up' to
encourage the child to draw strength from within themselves as well as those around
them. Writing prompts throughout the book help the child cope during their diagnosis
and express their thoughts and feelings during this dicult time. Examples of writing
prompts include: I am special because ...; When I'm bored in the hospital my family
and I ...; When I'm feeling sad it helps to ... etc. Free to children with cancer.
Romain, Trevor. Lift Me Up. American Childhood Cancer Organization, 2008. is 24
page book with inspirational text is lled with wonderful illustrations to color. Free
to children with cancer.
Grief in school
Gliko-Braden, Majel. Grief Comes to Class: A Teacher’s Guide. Centering Corporation, 1531
N. Saddle Creek Rd., Omaha, NE 68104. (402) 553-1200. Comprehensive guide
to grief in the classroom. Includes chapters on grief responses, the bereaved student,
teen grief, developmental changes, sample letter to parents, and sample teacher/parent
344
Appendix C: Resources
345
Appendix C: Resources
conferences. Also available on Amazon.com
e Compassionate Friends. Suggestions for Teachers and School Counselors. P.O. Box 3696,
Oak Brook, IL 60522. (630) 990-0010.
Romain, Trevor. What on Earth Do You Do When Someone Dies? Minneapolis, MN: Free
Spirit Publishing, 1999. Warm, honest words and beautiful illlustrations help children
understand and cope with grief.
Harvey, Diane. Why the Snowman Melts. Sandstone Publishing Saint George, UT, 2010.
rough the story of the melting snowman, young children gain understanding of
change and loss.
Brown, Laurie Krasny and Brown, Marc. When Dinosaurs Die: A Guide to Understanding
Death. Little Brown and Company, New York, NY, 1996. For children ages 5 to 8
years. Direct answers to children's questions about death such as "Why does someone
die?" Available through Amazon.com.
Wilhelm, Hans. I'll Always Love You. Crown Publishers, 1985. e loving story of a little
boy and his love and loss of his dog Ele. Available on Amazon.com.
Heckert, Connie. Dribbles. Clarion Books, New York, NY, 1994. For ages 5 to 8. is
picture book addresses death as told by three household cats living with an aging
owner and aging feline. Available on Amazon.com.
Grootman, Marilyn. When a Friend Dies: A Book for Teens about Grieving and Healing. Free
Spirit Publishing, Minneapolis, MN, 1994. Practical guide for teens that addresses
and validates emotions associated with death such as guilt, fear, anger and confusion.
Wolfelt, Alan. Healing a Child's Grieving Heart: 100 Practical Ideas for Families, Friends and
Caregivers. Companion Press, Ft. Collins, CO, 2001. Practical guide that provides
suggestions on how to help those who are grieving by oering sensitive responses
to "what to say and do" and "what not to say and do." Available on Amazon.com.
Hearing loss
Poitras Tucker, Bonnie. IDEA Advocacy for Children Who Are Deaf or Hard of Hearing: A
Question and Answer Book for Parents and Professionals. Singular Publishing Group,
1997.
IEP school advocacy
Siegel, Lawrence. e Complete IEP Guide: How to Advocate for Your Special Ed Child.
Harbor House Law Press, 2001. Spells out the IEP process for families and includes
helpful sample letters and forms.
Anderson, Winifred, Stephen Chitwood, and Deidre Hayden. Negotiating the Special
Education Maze: A Guide for Parents and Teachers. 3rd ed. Bethesda, Maryland:
Woodbine House, 1997. Excellent, well-organized text clearly explains the step-by-
step process necessary to obtain help for your child.
Susan Gorn, Editor. Special Education Dictionary. LRP Publications, 1997. (215) 784-0860.
Wright, Peter, and Wright, Pamela. Wrightslaw: Special Education Law. Harteld, VA:
Harbor House Law Press, 1999. Text of key laws and regulations.
Wright, Peter, and Wright, Pamela. Wrightslaw: From Emotions to Advocacy: e Special
Education Survival Guide. Harteld, VA: Harbor House Law Press, 2001. Full of
information on special education law, advocacy tactics, and IEP tips.
Speech and language
McAleer Hamaguchi, Patricia. Childhood Speech, Language and Listening Problems: What
Every Parent Should Know. John Wiley & Sons Inc., 1995.
Schoenbrodt, Lisa, ed. Children with Traumatic Brain Injury: A Parent’s Guide. Woodbine
House, 2003.
Schwartz, Sue, and Joan E. Heller Miller. e New Language of Toys: Teaching Communication
Skills to Special-Needs Children. Rockville, MD: Woodbine House, 1996.
Videotapes
Paul and the Dragon. Powerful 25 minute video created to help children, siblings and
friends understand the world of childhood cancer in a safe way, with humor but also
with truth. rough watching Paul’s battle with his dragon, the child with cancer will
understand that scary things will happen to them as they ght their "cancer-dragon."
ey will learn that the doctors, nurses and even the blue "medication-men" and
purple "chemo-blobs" are there to help them beat their cancer. Available through the
American Childhood Cancer Organization. http://www.acco.org
Why, Charlie Brown, Why? Tender story of a classmate who develops leukemia. Available as
a book or videotape. For video availability, call the Leukemia and Lymphoma Society,
(800) 955-4572 (4LSA).
Cancervive Back to School Kit. A comprehensive package of materials developed to assist
children and adolescents re-entering the school setting. e kit contains a “Teachers
Guide for Kids with Cancer” and two award-winning documentary videos: “Emily’s
Story: Back to School After Cancer” and “Making the Grade: Back to School
After Cancer for Teens.” http://www.cancersourcekids.com/parents/schoolintro.
cfm?usertypeid=3
Drying eir Tears. Produced by CARTI. For information, call (800) 482-8561. Video and
manual to help counselors, teachers, and other professionals help children deal with
the grief, fear, confusion and anger that occur after the death of a loved one. Has three
segments: one about training facilitators, one for children ages 5 to 8, and one for ages
9 to teens. Each section includes interviews with children and video from children’s
workshops. http://www.hopkinschildrens.org/tpl_rlinks_nobanner.aspx?id=828
Back to School: Teens Prepare for School Re-entry. Produced by Starbright Videos with
Attitude, call (800) 315-2580. Teens who have been there share their stories and
advice on how to get back into the groove of school. Also discusses how teens can
get the extra help they may need to make returning to school a successful experience.
http://www.starbright.org
Organizations
ACCO would like to acknowledge the many non-prot organizations that have been
founded in memory of children diagnosed with DIPG. It is because of the work of so many
of these organizations that DIPG has received increased awareness as well as heightened
research interest. e following is a list of organizations that support DIPG research
and/or provide resources to families on a national basis, whether nancial, emotional, or
informational. It is provided as a starting point to assist families, and is not to be regarded
as a comprehensive list.
346
Appendix C: Resources
347
Appendix C: Resources
Air Care Alliance
Email: [email protected]
(888) 260-9707
http://www.aircareall.org
e Air Care Alliance promotes, and provides public benet ying through facilitation of
ights for health, compassion and community service. ese groups do not y patients or
supplies when insurance or other funds can provide commercial transport via air ambulance,
charter or airline. e public benet ying volunteers y when nancial need or other
special circumstances mean a compelling human need would go unfullled.
American Childhood Cancer Organization®
10920 Connecticut Ave. Suite A
Kensington, MD 20895
(855) 858-2226
http://www.acco.org
Founded in 1970, ACCO has more than 70,000 members. Some of the free services
provided by ACCO include a toll-free information phoneline, an e-bulletin, childhood
cancer books to help children with cancer and their families, diagnosis kits, local support
group aliates, and national advocacy.
American Speech-Language-Hearing Association
2200 Research Blvd.
Rockville, MD 20850
(800) 638-8255; TTY: (301) 296-5650
http://www.asha.org
ASHA’s mission is to ensure that all people with speech, language and hearing disorders
have access to quality services to help them communicate eectively. Canadian organization
can be found at http://www.caslpa.ca
Believe in Tomorrow Children's Foundation
6601 Frederick Road
Baltimore, MD 21228
(800) 933-5470
http://www.believeintomorrow.org
e Believe in Tomorrow Children's Foundation provides hospital and respite housing
services to critically ill children and their families.
Brain Tumor Foundation of Canada
620 Colborne Street, Suite 301
London, ON N6B 3R9
(800) 265-5106; 519-642-7755
http://www.braintumour.ca
e Brain Tumor Foundation of Canada provides up-to-date brain tumor information
materials, educational events and support groups. Important brain tumor research is
supported through annual grants, a fellowship and the brain tumor tissue bank.
Childhood Brain Tumor Foundation
20312 Watkins Meadow Drive
Germantown, MD 20876
(877) 217-4166
http://www.childhoodbraintumor.org
Founded in 1994, the Childhood Brain Tumor Foundation funds scientic and clinical
research for pediatric brain tumors, and sponsors educational conferences.
Children's Brain Tumor Foundation (CBTF)
274 Madison Ave. Suite 1004
New York, NY 10016
(866) 228-HOPE
http://www.cbtf.org
Founded in 1988, the CBTF provides information, support and advocacy to children with
brain tumors and their families. ey fund scientic research leading to better treatments
and cures of pediatric brain tumors, as well as research leading to improved quality of life.
Compassionate Friends
900 Jorie Blvd. Suite 78
Oak Brook, IL 60523
(877) 969-0010
http://www.compassionatefriends.org
Compassionate Friends provides personal comfort, hope and support through local chapters
for bereaved family members experiencing the death of a child.
Just One More Day
1853 Surrey Court
Viera, FL 32955
(321) 698-8538
http://www.justonemoreday.org
Just One More Day is committed to providing information and support for families
aected by diuse intrinsic pontine glioma, promoting awareness, and funding research
leading to a cure.
Kids v Cancer
4646 Hawthorne Lane
Washington, DC 20016
(646) 361-3590
http://www.kidsvcancer.org
Kids v Cancer is committed to the creation of legislative initiatives leading to pediatric
cancer drug development, as well as the parent-led eort to making autopsy tissue donations
more widely available for research.
Make-A-Wish Foundation®of America
4742 N. 24th Street, Suite 400
Phoenix, AZ 85016
(800) 722-9474
http://www.wish.org
Founded in 1980, the Make-A-Wish Foundation has enriched the lives of children with
life-threatening medical conditions, and their families, through their wish-granting program.
National Association of Hospital Hospitality Houses (NAHHH)
P.O. Box 1439
Gresham, OR 97030
(800) 542-9730
http://www.nahhh.org
348
Appendix C: Resources
349
Appendix C: Resources
NAHHH is a nation-wide association of 200 non-prot organizations that provide lodging
and support services to patients and their families who are receiving medical treatment far
from their home communities.
Pediatric Brain Tumor Foundation
302 Ridgeeld Court
Asheville, NC 28806
(800) 253-6530
http://www.pbtfus.org
e PBTF, founded in 1991, provides education and emotional support for children with
brain tumors and their families. ey seek to nd the cause and cure for childhood brain
tumors by supporting medical research and increasing public awareness of childhood brain
tumors.
Reections of Grace Foundation
P.O. Box 298
Irwin, PA 15642
Email: contact@reectionsofgrace.org
http://www.reectionsofgrace.org
Reections of Grace Foundation is dedicated to providing nancial, emotional and
educational support for children and families ghting pediatric brain tumors; and funding
the search for a cure for DIPG and other forms of pediatric brain tumors.
Smiles for Sophie Forever Foundation
31722 Leeward Court
Avon Lake, OH 44012
Email: info@smilesforsophieforever.org
http://www.smilesforsophieforever.org
Smiles for Sophie Forever is dedicated to providing nancial support to families burdened
by pediatric brain tumors, as well as increasing global awareness of the devastation of
pediatric brain tumors.
SuperSibs!
660 N. First Bank Drive
Palatine, IL 60067
(888) 417-4704
http://www.supersibs.org
SuperSibs' mission is to support, honor and recognize brothers and sisters of children
with cancer. ey provide numerous “Advocacy and Support” services, including journals
for siblings, guides, scholarships and a monitored teen chat internet room. SuperSibs also
sponsors “Surprise and Delight” services such as special sibling activities and giveaways for
siblings ages 4 to 18. Services are provided free of charge.
e Cure Starts Now Foundation
10280 Chester Road
Cincinnati, OH 45215
(513) 772-4888
http://www.thecurestartsnow.org
e Cure Starts Now Foundation ghts for the cure for children with brainstem glioma
through public awareness and media campaigns, as well as the funding of research leading
to new treatments for DIPG.
Online Support Groups
ACOR, e Association of Cancer Online Resources, Inc.
http://www.acor.org
ACOR oers access to mailing lists that provide support, information, and community to everyone
aected by cancer and related disorders. It hosts numerous pediatric cancer discussion groups.
American Childhood Cancer Organization's Inpire Online Community
http://www.inspire.com/groups/american-childhood-cancer-organization
ACCO's Inspire online commmunity connects patients, families, friends and caregivers.
It provides a platform for support and inspiration from diagnosis, through treatment and
beyond. Discussion topics include: newly diagnosed; treatment; emotional support for
children with cancer, siblings, parents and caregivers; nancial and insurance issues, and
more.
Apraxia-Kids Mailing List
Listserv@Listserv.syr.edu
http://www.apraxia-kids.org
is website and mailing list covers oral motor apraxia and related disabilities. To subscribe,
send an email with the message “subscribe apraxia-kids FirstName LastName.”
Cerebellar Mutism Brain Tumor Listserv Yahoogroup
http://health.groups.yahoo.com/group/cerebellarmutism
Listserv providing online support for parents and caregivers of children who suer from
cerebellar mutism and posterior fossa syndrome after brain tumor surgery/resection.
DIPG Listserv Yahoogroup
http://health.groups.yahoo.com/group/dipg
is group is primarily for parents of children diagnosed with diuse intrinsic pontine
glioma (DIPG). e membership includes parents who are in all stages of the DIPG journey.
Educating Brain Tumor Kids
http://groups.yahoo.com/group/EducatingBTKids
A group with links and les dealing with neuropsychological testing, school re-entry, school
options, late eects etc. ere is an associated listserv with archives.
Home Schooling Special Needs Children
http://groups.yahoo.com/group/special-needs-homeschool
is group supports parents who choose to home school their children with special needs.
Most members have medically fragile children dealing with challenges in speech, motor
development and learning disabilities and home school full time or part of the time.
Hydrocephalus (HYCEPH_L)
http://neurosurgery.mgh.harvard.edu/pedi/hyceph-l.htm
is list is open to all people interested in hydrocephalus.
IEP Guide and Listserv Yahoogroup
http://groups.yahoo.com/group/IEP_guide
350
Appendix C: Resources
351
Appendix C: Resources
is is a very large listserv that oers special education support and has a free IEP guidebook.
Pediatric Brain Tumor Angels Listserv Yahoogroup
http://health.groups.yahoo.com/group/PBTAngels
Listserv providing online support for parents and caregivers who are facing end of life issues
with a child who has a brain tumor and extended support for parents of children who have
died after battling a brain tumor.
Pediatric Brain Tumor Facial Paralysis Listserv Yahoogroup
http://health.groups.yahoo.com/group/PBTFacialParalysis
Listserv providing online support for parents and caregivers to gain information and support
regarding facial nerve paralysis after surgery for pediatric brain tumor surgery/resection.
Pediatric Brain Tumor Listserv Yahoogroup
www.yahoogroups.com/group/pediatricbraintumors
Listserv providing information and online support for parents and caregivers of children
diagnosed with pediatric brain tumors including: astrocytoma, atypical teratoid/
rhabdoid, glioblastoma multiforme, pleomorphic xanthoastrocytoma, craniopharyngioma,
diuse intrinsic pontine glioma, gangliocytoma, ganglioglioma, germinoma, glioma,
medulloblastoma, metastatic brain tumor, neurocytoma, oligodendroglioma, juvenile
pilocytic astrocytoma, pineocytoma, pineoblastoma, PNET, primitive neuroectodermal
tumor, teratoma, and ependymoma.
Websites
Bandaids and Blackboards-When Chronic Illness Goes to School
http://www.lehman.cuny.edu/faculty/jeitas/bandaides/contkids.html
Wonderful, fun and informative website about ill children and school.
CaringBridge
http://www.caringbridge.org
Free, personal and private websites to help families experiencing a health crisis connect
with family and friends.
Children's Hospice and Palliative Care Coalition
http://www.childrenshospice.org/coalition
Children’s Hospice & Palliative Care Coalition is a social movement led by children’s
hospitals, hospices, home health and grassroots agencies and individuals to improve care
for children with life-threatening conditions and their families.
Children's Oncology Camping Association International
http://www.cocai.org
Website listing the more than 65 children's oncology camps located across the U.S. as well
as camps for children with cancer in Canada, New Zealand and Europe.
Children's Oncology Group
http://www.childrensoncologygroup.org
e Children's Oncology Group (COG) unites more than 7,500 experts in over 200
children's hospitals, universities and cancer centers into a global team dedicated to working
towards a cure for all children with cancer. Includes a list of all COG treatment centers.
Clinical Trials.gov
http://www.clinicaltrials.gov
Clinical Trials.gov is a registry and results database of federally and privately supported
clinical trials conducted in the U.S. and around the world.
DIPG Collaborative
http://dipg.org
DIPG Collaborative provides information on numerous foundations whose mission is to
support DIPG research and support children and families diagnosed with DIPG.
DIPG Registry
http://dipgregistry.org
Comprehensive website dedicated solely to DIPG. Divided into two portals, one for patients
and their families, and one for medical professionals. Information includes upcoming
conferences, DIPG research studies, available clinical trials, registry enrollment form, up-
to-date information on the diagnosis and treatment of DIPG as well as contact information
for DIPG specialists in Canada, U.S., Australia and Europe, providing second opinions.
iCANcer Electronic Medical Record
http://itunes.apple.com/us/app/icancer/id389815342?mt=8
Personal electronic medical record created for iPhone, iPod, and/or iPad app that stores
diagnosis, treatment and health care provider information. is app manages medical
information including current and past medications, side eects, lab results (graphed over
time), and it organizes and syncs doctor's appointments, and conveniently exports medical
information to an email format for easy communication to a health care provider prior
to an appointment.
Monkey In My Chair
http://www.monkeyinmychair.org
Monkey In My Chair is a program for preschool and elementary aged children who are
away from school because of a cancer diagnosis. Each child is provided with a "monkey
kit" which includes a teacher's guide and classroom book, a backpack and a big stued
monkey that takes the child's place when he/she is unable to be in school.
Pediatric Brain Tumor Consortium (PBTC)
http://www.pbtc.org
e PBTC is a multidisciplinary cooperative research organization devoted to the study
of correlative tumor biology and new therapies for primary CNS tumors of childhood.
Pediatric Preclinical Testing Initiative
http://pptiohsu.blogspot.com/2011/12/open-science-forum-dipg-preclinical.html
Research blogspot including postings on the Rapid Preclinical Development of Targeted
erapy Combinations for DIPG.
Autopsy tissue and organ donation
Oregon Health and Science University CCURE-FAST
352
Appendix C: Resources
353
Appendix C: Resources
http://www.ohsu.edu/xd/health/services/doernbecher/research-education/research/pape-
family-pediatric-research-institute/ccure-fast.cfm
A helpful site to assist families with information about tumor tissue donation. Includes
dowloadable information on tumor banking, Q&A about legacy gifting, religion and tumor
banking, as well as guidelines for medical professionals.
U.S. Government Information on Organ and Tissue Donation and Transplantation
http://www.organdonor.gov
Comprehensive website dedicated to providing information on organ and tissue donation
and transplantation, including statistics, information on how to become an organ donor,
legislation, associated research and grant opportunities.
Trillium Gift of Life Network
http://www.giftoife.on.ca/en/organandtissuedonation
Website to assist Canadian families with information about organ and tissue donation.
Behavior
Behavior Problems of Children who have undergone Treatment for Brain Tumors
http://www.childhoodbraintumor.org/index.php?option=com_content&view=articl
e&id=59:behavior-problems-in-children-who-have-undergone-treatment-for-brain-
tumors&catid=38:late-eects-a
Cerebellar mutism
Cerebellar Mutism and Posterior Fossa Syndrome
http://groups.yahoo.com/group/cerebellarmutism
ere is an annotated bibliography from a medline search on cerebellar mutism, a
bibliography on radiation and cognitive eects and several articles from a speech pathologist.
Disabilities
Council of Educators for Students with Disabilities, Inc. (CESDI)
http://www.504idea.org/Council_Of_Educators/Welcome.html
CESDI provides Section 504 and special education training and resources to educators.
Protection and Advocacy
http://www.disabilityrightsca.org
Group that works to advance the human and legal rights of people with disabilities. Website
includes a page on assistive technologies.
Pacer Center Parent Advocacy Coalition for Educational Rights
http://www.pacer.org
A national coalition of parents working for educational rights.
Family Village: A Global Community of Disability Resources
http://www.familyvillage.wisc.edu
A huge site that provides informational resources on specic diagnoses, communication
connections, adaptive products and technology, adaptive recreational activities, education,
health issues, disability-related media and literature.
Distance learning
Talia Seidman Foundation
http://www.taliaseidman.com
An organization dedicated to using technology to bring hospitalized and homebound
chronically ill children back into the classroom.
Hearing impairment
Hard of Hearing & Deaf Students: Resource Guide to Support Classroom Teachers
http://www.bced.gov.bc.ca/specialed/hearimpair/intro.htm
Homeschooling
A to Z’s Cool Homeschooling
http://www.gomilpitas.com/homeschooling
Huge site with information on introduction to home schooling, curricula, home schooling
laws, support groups, methods, and philosophies.
National Home Education Network
http://www.homeschool-curriculum-and-support.com/national-home-education-network.html
A source for home schooling information, support group listings, home school news, and
related resources.
Nonverbal learning disabilities
Nonverbal Learning Disabilities
http://www.nldontheweb.org
A comprehensive site on nonverbal learning disabilities.
Siblings
SuperSibs
http://www.supersibs.org
A national program dedicated to the interests of brothers and sisters of children with cancer.
Includes online activities for children at http://supersibs.org/the-sib-spot/index.html.
Siblings of People with Disabilities
http://www.iidc.indiana.edu/index.php?pageId=2458
A list of books and videos that help siblings of people with disabilites.
Special education law
Consortium for Appropriate Dispute Resolution in Special Education (CADRE)
http://www.directionservice.org/cadre
CADRE provides support and materials that can help parents and educators implement
354
Appendix C: Resources
the mediation requirements under IDEA 97.
Department of Education Information about IDEA
http://idea.ed.gov
Comprehensive information about the Individuals with Disabilities Education Act.
Wrights Special Education Law
http://www.wrightslaw.com
is extensive and well organized site is probably the best place to start to gather information
on special education law. Includes sections on advocacy, law, books and other resources.
Educational Rights/Educational Law
http://edlaw.org/wordpress
is site provides publications and services for attorneys, advocates and parents who need
to know about educational law, including a section that deals with transportation.
Americans with Disabilities Act Homepage
http://www.usdoj.gov/crt/ada/adahom1.htm
National Information Center for Children and Youth with Disabilities
http://www.nichcy.org
Includes helpful resource sheets for every state.
Speech and language
IntelliTools
http://www.intellitools.com
is rm has a great catalog of assistive technology and communication devices.
Sports
Disabled Sports USA
http://www.dsusafw.org
An organization that gives people with physical, neuromuscular and developmental
impairments the opportunity to participate in a variety of activities including water/snow
skiing, camping, and whitewater rafting. Adaptive equipment information available.
American Hippotherapy Association
http://www.americanequestrian.com/hippotherapy.htm
Hippotherapy is therapeutic riding for those with motor disturbances.
North American Riding for the Handicapped
http://www.narha.org
An organization that promotes the benets of horseback riding for those with physical,
emotional or learning disabilities.
355
Appendix D: Research Articles
Appendix D
Research Articles
e following journal articles were referenced by the authors in specic chapters
throughout the book. ey are listed here to provide direction for additional
reading for those individuals who wish to delve deeper into a specic topic.
Further Reading
References from Chapter 4, Imaging DIPG
1) Jallo GI, Biser-Rohrbaugh A, Freed D, (2004). Brainstem Gliomas. Childs Nervous
System, 20: 143-153.
2) Barkovich AJ, Krischer J, Kun LE, et al., (1990). Brain Stem Gliomas: A Classication
System Based on Magnetic Resonance Imaging. Pediatric Neurosurgery, 16: 73-83.
3) Moghrabi A, Kerby T, Tien RD, et al., (1995). Prognostic Value of Contrast-Enhanced
Magnetic Resonance Imaging in Brainstem Gliomas. Pediatric Neurosurgery, 23: 293-298.
4) Fischbein NJ, Prados MD, Wara W, et al., (1996). Radiographic Classication of Brainstem
Tumors: Correlation of Magnetic Resonance Imaging Appearance with Clinical Outcome.
Pediatric Neurosurgery 24: 9-23.
5) Freeman CR, Farmer JP, (1998). Pediatric Brain Stem Gliomas: A Review. Int. J. Radiation
Oncology Biol Phys 40(2): 265-271.
6) Fisher PG, Breiter SN, Carson BS, et al., (2000). A Clinicopathologic Reappraisal of Brain
Stem Tumor Classication: Identication of Pilocytic Astrocytoma and Fibrillary Astrocytoma
as Distinct Entities. Cancer 89(7): 1569-1576.
7) Hargrave D, Chuang N, Bouet E, (20 08). Conventional MRI Cannot Predict Survival
in Childhood Diuse Intrinsic Pontine Glioma. J. Neurooncol 86: 313-319.
8) Liu AK, Brandon J, Foreman NK, et al., (2009). Conventional MRI at Presentation Does
Not Predict Clinical Response to Radiation erapy in Children with Diuse Pontine Glioma.
Pediatr Radiol 39: 1317-1320.
9) Kornreich L, Schwarz M, Karmazyn B, et al., (2005). Role of MRI in the Management of
Children with Diuse Pontine Tumors: A Study of 15 Patients with Review of the Literature.
Pediatric Radiology 35: 872-879.
10) Nelson MD, Soni D, Baram TZ, (1994). Necrosis in Pontine Gliomas: Radiation Induced
or Natural History? Radiology 191: 279:282.
11) Fenton LZ, Madden JR, Foreman NK, (2003). Misleading Leads: Brain Stem Glioma in a
Child: False Diagnosis of Radiation Necrosis With FDG PET. Med Pediatr Onc 40: 260–262.
12) Krieger MD, Bluml S, McComb JG, (2003). Magnetic Resonance Spectroscopy of Atypical
Diuse Pontine Masses. Neurosurg Focus 15(1): 1-4.
356
Appendix D: Research Articles
357
Appendix D: Research Articles
13) Panigrahy A, Nelson MD, Finlay JL, et al., (2008). Metabolism of diuse intrinsic
brainstem gliomas in children. Neuro-Oncology 10 (1): 32-44.
14) Mullins ME, (2006). MR Spectroscopy: Truly Molecular Imaging; Past, Present and
Future. Neuroimag Clin N Am 16: 605–618.
15) Curless RG, Bowen BC, Pattany PM, et al., (2002). Magnetic Resonance Spectroscopy
in Childhood Brainstem Tumors. Pediatric Neurology 26(5): 374-378.
16) Smith JK, Londono A, Castillo M, et al., (2002). Proton Magnetic Resonance Spectroscopy
of Brainstem Lesions. Neuroradiology 44: 825-829.
17) Panigrahy A, Bluml S, (2009). Neuroimaging of Pediatric Brain Tumors: From Basic to
Advanced Magnetic Resonance Imaging. Journal of Child Neurology 24(11): 1343-1365.
18) Laprie A, Pirzkall A, Hass-Kogan DA, et al., (2005). Longitudinal Multivoxel MR
Spectroscopy Study of Pediatric Diuse Brainstem Gliomas Treated with Radiotherapy. Int. J.
Radiation Oncology Biol. Phys. 62(1): 20-31.
19) Law M, Oh S, Johnson G, et al., (2006). Perfusion Magnetic Resonance Imaging Predicts
Patient Outcome as an Adjunct to Histopathology: A Second Reference Standard in the Surgical
and Nonsurgical Treatment of Low-Grade Gliomas. Neurosurgery 58(6): 1099-1107.
20) Bisdas S, Kirkpatrick M, Giglio P, et al., (2009). Cerebral Blood Volume Measurements by
Perfusion-Weighted MR Imaging in Gliomas: Ready for Prime Time in Predicting Short-Term
Outcome and Recurrent Disease? Am J. Neuroradiol 30: 681-688.
21) Law M, Young RJ, Babb JS, et al., (2008). Gliomas: Predicting Time to Progression
or Survival with Cerebral Blood Volume Measurements at Dynamic Susceptibility-weighted
Contrast-enhanced Perfusion MR Imaging. Radiology 247(2): 490-498.
22) Cha S, (2003). Perfusion MR imaging: Basic principles and clinical applications. Magn
Reson Imaging Clin N Am 11: 403–413.
23) Macapinlac HA, (2006). Positron Emission Tomography of the Brain. Neuroimag Clin
N Am 16: 591–603.
24) Pirotte BJM, Lubansu A, Massagner N, et al., (2007). Results of positron emission
tomography guidance and reassessment of the utility of and indications for stereotactic biopsy in
children with inltrative brainstem tumors. J. Neurosurg 107(5 Suppl Pediatrics): 392–399.
25) Kwon JW, Kim IO, Cheon JE, et al., (2006). Paediatric brain-stem gliomas: MRI, FDG-
PET and histological grading correlation. Pediatr Radiol 36: 959–964.
References from Chapter 8, Radiosensitizers for DIPG
1) Shrieve DC, Loeer JS, (1995). Advances in radiation therapy for brain tumors. Neurol
Clin 13(4): 773-793.
2) Stieber VW, Mehta MP, (2007). Advances in radiation therapy for brain tumors. Neurol
Clin 25: 1005-1033.
3) Sanghavi SN, Needle MN, Krailo MD, Geyer JR, Ater J, Mehta MP, (2003). A phase
1 study of topotecan as a radiosensitizer for brainstem glioma of childhood: First report of the
Children’s Cancer Group-0952. Neuro Oncol 5(1): 8-13.
4) Packer RJ, Krailo M, Mehta M, Warren K, Allen J, Jakacki R, Villablanca JG, Chiba A,
Reaman G, (2005). Phase 1 study of concurrent RMP-7 and carboplatin with radiotherapy
for children with newly diagnosed DIPGs. Cancer 104(6): 1281-1287.
5) Turner CD, Chi S, Marcuc KJ, MacDonald T, Packer RJ, Poussaint TY, Vajapeyarn S,
Ullrich N, Briody C, Chordas C, Zimmerman MA, Kieran M, (2007). Phase II study of
thalidomide and radiation in children with newly diagnosed brain stem gliomas and glioblastoma
multiforme. Journal of Neuro-Oncology 82: 95-101.
6) Packer RJ, Prados M, Phillips P, Nicholson HS, Boyett JM, Goldwein J, Rorke LB,
Needle MN, Sutton L, Zimmerman RA, Fitz CR, Vezina LG, Etcubanas E, Wallenberg
JC, Reman G, Wara W, (1996). Treatment of children with newly diagnosed brain stem
gliomas with intravenous recombinant B-interferon and hyperfractionated radiation therapy.
A Children’s Cancer Group Phase I/II study. Cancer 77(10): 2150-2156.
References from Chapter 9, Chemotherapy and Biologics
1) Jenkin RDT, Boesel C, Ertel I, et al., (1987). Brain-stem tumors in childhood: a prospective
randomized trial of irradiation with and without adjuvant CCNU, VCR, and prednisone. J.
Neurosurg 66: 227-233.
2) Walter AW, Gajjar A, Ochs SJ, et al., (1998). Carboplatin and etoposide with
hyperfractionated radiotherapy in children with newly diagnosed diuse pontine gliomas: a
phase I/II study. Med Ped Oncol 30: 28-33.
3) Chamberlain MC., (2993). Recurrent brainstem glioma treated with oral VP-16. J.
Neurooncol15: 133-139.
4) Korones DN, Fisher PG, Kretschmar C, et al., (2008). Treatment of children with diuse
intrinsic brain stem glioma with radiotherapy, vincristine and oral VP-16: a Children’s Oncology
Group Phase II study. Pediatr Blood Cancer 50: 227-230.
5) Dreyer ZE, Kadota RP, Stewart CF, et al., (2003). Phase 2 study of idarubicine in pediatric
brain tumors: Pediatric Oncology Group study POG 9237. Neuro-oncology 5: 261-267.
6) Wol JEA, Westphal S, MolenkampG, et al., (2002).Treatment of paediatric pontine
glioma with oral trophosphamide and etoposide. British Journal of Cancer 87: 945-949.
7) Wol JEA, Driever PH, Erdlenbruch B, et al., (2010). Intensive chemotherapy improves
survival in pediatric high-grade glioma after gross total resection: results of the HIT-gBM-c
protocol. Cancer 116: 705-712.
8) Minturn JE, Janss AJ, Fisher PG, et al., (2011). A phase II study of metronomic oral
topotecan for recurrent childhood brain tumors. Pediatr Blood Cancer 56: 39-44.
9) Broniscer A, Iacono L, Chintagumpala M, et al., (2005). Role of temozolomide after
radiotherapy for newly diagnosed diuse brainstem glioma in children. Cancer 103: 133-139.
10) Cohen KJ, Heideman RL, Zhou T, et al., (2011). Temozolomide in the treatment of
children with newly diagnosed diuse intrinsic pontine gliomas: a report from the Children's
Oncology Group. Neuro-Oncol 13:410-416.
11) Chiang KL, Chang KP, Lee YY, et al., (2010). Role of temozolomide in the treatment of
newly diagnosed diuse brainstem glioma in children: experience at a single institution. Childs
Nerv Syst 26: 1035-1041.
12) Jalali R, Raut N, Arora B, et al., (2010) Prospective evaluation of radiotherapy with
concurrent and adjuvant temozolomide in children with newly diagnosed diuse intrinsic
pontine glioma. Int J. Radiation Oncology Biol Phys 77: 113-118.
13) Sharp JR, Bouet E, Stempak D, et al., (2010). A multi-center Canadian pilot study of
metronomic temozolomide combined with radiotherapy for newly diagnosed paediatric brainstem
glioma. European Journal of Cancer 46: 3271-3279.
14) Sirachainan N, Pakakasama S, Visudithbhan A, et al., (2008). Concurrent radiotherapy
358
Appendix D: Research Articles
359
Appendix D: Research Articles
with temozolomide followed by adjuvant temozolomide and cis-retinoic acid in children with
diuse intrinsic pontine glioma. Neuro-Oncology 10: 577-582.
15) Kim CY, Kim SK, Phi JH, et al., (2010). A prospective study of temozolomide plus
thalidomide during and after radiation therapy for pediatric diuse pontine gliomas: preliminary
results of the Korean Society for Pediatric Neuro-Oncology study. J. Neurooncol 100: 193-198.
16) Kretschmar CS, Tarbell NJ, Barnes PD, Krischer JP, Burger PC, Kun L., (1992). Pre-
irradiation chemotherapy and hyperfractionated radiation therapy 66Gy for children with brain
stem tumors. Cancer 72: 1404-1413.
17) Jennings MT, Sposto R, Boyett JM, et al., (2002). Preradiation chemotherapy in primary
high-risk brainstem tumors: phase II study CCG-9941 of the Children’s Cancer Group. J. Clin
Oncol 20: 3431-3437.
18) Doz F, Neuenschwander S, Bouet E, et al., (2002). Carboplatin before and during
radiation therapy for the treatment of malignant brain stem tumours: a study by the Societe
Francaise d’Oncologie Pediatrique. European Journal of Cancer 38: 815-819.
19) Graham ML, Herndon JE, Casey JR, et al., (1997). High-dose chemotherapy with
autologous stem-cell rescue in patients with recurrent and high-risk pediatric brain tumors. J.
Clin Oncol 15: 1814-1823.
20) Finlay JL, Goldman S, Wong MC, et al., (1996). Pilot study of high-dose thiotepa and
etoposide with autologous bone marrow rescue in children and young adults with recurrent CNS
tumors. J. Clin Oncol 14: 2495-2503.
21) Bouet E, Raquin M, Doz F, et al., (2000). Radiotherapy followed by high dose busulfan
and thiotepa. Cancer 88: 685-692.
22) Bouet E, Khelfasoui F, Philip I, Biron P, Brunat-Mentigny M, Phlip T., (1997).
High-dose carmustine for high-grade gliomas in childhood. Cancer Chemother Pharmacol
39: 376-379.
23) Jakacki RI, Siert J, Jamison C, Velasquez L, Allen JC., (1999). Dose-intensive, time-
compressed procarbazine, CCNU, vincristine (PCV) with peripheral blood stem cell support
and concurrent radiation in patients with newly diagnosed high-grade gliomas. J. Neuro-Oncol
44: 77-83.
24) Kedar A, Maria BL, Graham-Pole J, et al., (1994). High dose chemotherapy with marrow
reinfusion and hyperfractionated irradiation for children with high-risk brain tumors. Med
Ped Oncol 23: 428-436.
25) Dunkel IJ, Garvin JH, Goldman S, et al., (1998). High dose chemotherapy with
autologous bone marrow rescue for children with diuse pontine brain stem tumors. J. Neuro
Oncol 37:67-73.
26) Allen J, Packer R, Bleyer A, et al., (1991). Recombinant interferon beta: a phase I-II trial
in children with recurrent brain tumors. J. Clin Oncol 9: 783-788.
27) Packer RJ, Prados M, Phillips P, et al., (1996). Treatment of children with newly diagnosed
brain stem gliomas with intravenous recombinant beta-interferon and hyperfractionated
radiation therapy. Cancer 77: 2150-2156.
28) Packer RJ, Krailo M, Mehta M, et al., (2005). A phase I study of concurrent RMP-7
and carboplatin with radiation therapy for children with newly diagnosed brainstem gliomas.
Cancer 104: 1968-1974.
29) Hall WA, Doolittle ND, Daman M, et al., (2006). Osmotic blood-brain barrier disruption
chemotherapy for diuse pontine gliomas. J. Neuro-Oncol 77: 279-284.
30) Greenberg ML, Fisher OG, Freeman C, et al., (2005). Etoposide, vincristine, and
cyclosporine A with standard-dose radiation therapy in newly diagnosed diuse intrinsic brainstem
gliomas: a Pediatric Oncology Group Phase 1 study. Pediatr Blood Cancer 45: 644-648.
31) Gururangan S, Chi SN, Poussaint TY, et al., (2010). Lack of ecacy of bevacizumab
plus irinotecan in children with recurrent malignant glioma and diuse brainstem glioma: a
pediatric brain tumor consortium study. J. Clin Oncol 28: 3069-3075.
32) Gilbertson RJ, Hill DA, Hernan R, et al., (2003). ERBB1 is amplied and overexpressed
in high-grade diusely inltrative pediatric brain stem glioma. Clin Can Res 9: 3620-3624.
33) Bode U, Buchen S, Janssen G, et al., (2006). Results of a phase II trial of h-R3 monoclonal
antibody (nimotuzumab) in the treatment of resistant or relapsed high-grade gliomas in children
and adolescents. ASCO Meeting Abstracts: 1522
34) Geoerger B, Hargrave D, omas F, et al., (2011). Innovative therapies for children with
cancer pediatric phase I study of erlotinib in brainstem glioma and relapsing/refractory brain
tumors. Neuro-Oncology 13: 109-118.
35) Michalski A, Bouet E, Taylor RE, et al., (2010). e addition of high-dose tamoxifen
to standard radiotherapy does not improve the survival of patients with diuse intrinsic pontine
glioma. J. Neuro-Oncol 100: 81-88.
36) Burzynski SR, Janicki TJ, Weaver RA, Burzynski B., (2006). Targeted therapy with
antineoplastons A10 and AS2-1 of high-grade, recurrent, and progressive brainstem glioma.
Integrative Cancer erapies 5: 40-47.
References from Chapter 10, e Use of Steroids in Patients with DIPG
1) Ryan J., (Jan. 2000). Radiation somnolence syndrome. J. Pediatr Oncol Nurs 17(1): 50-53.
2) Dietrich J, Rao K, Pastorino S, Kesari S., (Mar 2011). Corticosteroids in brain cancer
patients: benets and pitfalls. Expert Rev Clin Pharmacol 4(2): 233-242.
3) Moliterno JA, Henry E, Pannullo SC., (Sept 2009). Corticorelin acetate injections for
the treatment of peritumoral brain edema. Expert Opin Investig Drugs 18(9): 1413-1419.
4) Liu AK, Macy ME, Foreman NK., (Nov 2009). Bevacizumab as therapy for radiation
necrosis in four children with pontine gliomas. Int J. Radiat Oncol Biol Phys 75(4):1148-1154.
References from Chapter 11, Caring for Your Child at Home
1) Aging with Dignity, (2010). Five Wishes. Retrieved from Aging with Dignity: http://
www.agingwithdignity.org/ve-wishes.php.
2) Aging wth Dignity, (2010). My Wishes. Retrieved from Aging with Dignity: http://www.
agingwithdignity.org/catalog/product_info.php?products_id=85.
3) Coda Alliance, (2011). Go Wish - Go Wish Interactive. Retrieved from e Go Wish
Game: http://www.gowish.org/staticpages/index.php/thegame.
4) Ethier, A, Rollins, J & Stewert, J. (Eds)., (2010). Pediatric Oncology Palliative and End-
of-life Care Resource. Glenview, IL, Association of Pediatric Oncology Nurses.
5) Kuttner, L., (1996). A Child in Pain: How to Help and What to Do. Point Roberts, WA,
Hartley & Marks.
6) Orlo, S., & Hu, S. M. (Eds.). (2003). Home Care for Seriously Ill Children: A Manual
for Parents. 3rd ed., Alexandria, Virginia, Children's Hospice International.
360
Appendix D: Research Articles
361
Appendix D: Research Articles
References from Chapter 14, Future of Genomics and Proteomics in DIPG
1) Albright AL, Packer RJ, Zimmerman R, Rorke LB, Boyett J, Hammond GD, (1993).
Magnetic resonance scans should replace biopsies for the diagnosis of diuse brain stem gliomas:
A report from the Children’s Cancer Group. Neurosurgery 33(6): 1026-1029; discussion
1029-1030.
2) Angelini P, Hawkins C, Laperriere N, Bouet E, Bartels U, (2010). Post mortem
examinations in diuse intrinsic pontine glioma: challenges and chances. J. Neurooncol
101(1): 75-81.
3) Badhe PB, Chauhan PP, Mehta NK, (2004). Brainstem gliomas: A clinicopathological
study of 45 cases with p53 immunohistochemistry. Indian J. Cancer 41(4): 170-174.
4) Barrow J, Adamowicz-Brice M, Cartmill M, Macarthur D, Lowe J, Robson K, Brundler
MA, Walker DA, Coyle B, Grundy R, (2010). Homozygous loss of ADAM3A revealed by
genome-wide analysis of pediatric high-grade glioma and diuse intrinsic pontine gliomas.
Neuro Oncol 13(2): 212-222.
5) Broniscer A, Baker JN, Baker SJ, Chi SN, Geyer JR, Morris EB, Gajjar A, (2010).
Prospective collection of tissue samples at autopsy in children with diuse intrinsic pontine
glioma. Cancer 116(19): 4632-4637.
6) Caretti V, Zondervan I, Meijer DH, Idema S, Vos W, Hamans B, Bugiani M, Hulleman
E, Wesseling P, Vandertop WP, Noske DP, Kaspers G, Moltho CF, Wurdinger T, (2011).
Monitoring of Tumor Growth and Post-Irradiation Recurrence in a Diuse Intrinsic Pontine
Glioma Mouse Model. Brain Pathol. 21(4): 441-451.
7) Chico-Ponce de Leon F, Perezpena-Diazconti M, Castro-Sierra E, Guerrero-Jazo FJ,
Gordillo-Dominguez LF, Gutierrez-Guerra R, Salamanca T, Sosa-Sainz G, Santana-Montero
BL, DeMontesinos-Sampedro A, (2003). Stereotactically-guided biopsies of brainstem tumors.
Childs Nerv Syst 19(5-6): 305-310.
8) Frazier JL, Lee J, omale UW, Noggle JC, Cohen KJ, Jallo GI, (2009). Treatment
of diuse intrinsic brainstem gliomas: failed approaches and future strategies. J. Neurosurg
Pediatr 3(4): 259-269.
9) Giese H, Homann KT, Winkelmann A, Stockhammer F, Jallo GI, omale UW,
(2010). Precision of navigated stereotactic probe implantation into the brainstem. J. Neurosurg
Pediatr 5(4): 350-359.
10) Hashizume R, Ozawa T, Dinca EB, Banerjee A, Prados MD, James CD, Gupta, N,
(2010). A human brainstem glioma xenograft model enabled for bioluminescence imaging. J.
Neurooncol 96(2): 151-159.
11) Jallo GI, Penno M, Sukay L, Liu JY, Tyler B, Lee J, Carson BS, Guarnieri M, (2005).
Experimental models of brainstem tumors: development of a neonatal rat model. Childs Nerv
Syst 21(5): 399-403.
12) Jallo GI, Volkov A, Wong C, Carson BS Sr, Penno MB, (2006). A novel brainstem tumor
model: functional and histopathological characterization. Childs Nerv Syst 22(12): 1519-1525.
13) Kumar HR, Zhong X, Sandoval JA, Hickey RJ, Malkas LH, (2008). Applications of
emerging molecular technologies in glioblastoma multiforme. Expert Rev Neurother 8(10):
1497-1506.
14) Kwon JW, Kim IO, Cheon JE, Kim WS, Moon SG, Kim TJ, Chi JG, Wang KC, Chung
JK, Yeon KM, (2006). Paediatric brain-stem gliomas: MRI, FDG-PET and histological grading
correlation. Pediatr Radiol 36(9): 959-964.
15) Laprie A, Pirzkall A, Haas-Kogan DA, Cha S, Banerjee A, Le TP, Lu Y, Nelson S,
McKnight TR, (2005). Longitudinal multivoxel MR spectroscopy study of pediatric diuse
brainstem gliomas treated with radiotherapy. Int J. Radiat Oncol Biol Phys 62(1): 20-31.
16) Leach PA, Estlin EJ, Coope DJ, orne JA, Kamaly-Asl ID, (2008). Diuse brainstem
gliomas in children: should we or shouldn't we biopsy? Br J Neurosurg 22(5): 619-624.
17) Lee J, Jallo GI, Guarnieri M, Carson BS Sr, Penno MB, (2005). A novel brainstem
tumor model: guide screw technology with functional, radiological, and histopathological
characterization. Neurosurg Focus 18(6A): E11.
18) Lonser RR, Warren KE, Butman JA, Quezado Z, Robison RA, Walbridge S, Schiman
R, Merrill M, Walker ML, Park DM, Croteau D, Brady RO, Oldeld EH, (2007). Real-
time image-guided direct convective perfusion of intrinsic brainstem lesions. Technical note.
J. Neurosurg 107(1): 190-197.
19) Mursch K, Halatsch ME, Markakis E, Behnke-Mursch J, (2005). Intrinsic brainstem
tumours in adults: results of microneurosurgical treatment of 16 consecutive patients. Br J.
Neurosurg 19(2): 128-136.
20) Paugh BS, Qu C, Jones C, Liu Z, Adamowicz-Brice M, Zhang J, Bax DA, Coyle B,
Barrow J, Hargrave D, Lowe, J, Gajjar A, Zhao W, Broniscer A, Ellison DW, Grundy RG,
Baker SJ, (2010). Integrated molecular genetic proling of pediatric high-grade gliomas reveals
key dierences with the adult disease. J. Clin Oncol 28(18): 3061-3068.
21) Perez-Gomez JL, Rodriguez-Alvarez CA, Marhx-Bracho A, Rueda-Franco F, (2010).
Stereotactic biopsy for brainstem tumors in pediatric patients. Childs Nerv Syst 26(1): 29-34.
22) Pichler R, Pichler J, Mustafa H, Nussbaumer K, Zaunmuller T, Topakian R, (2007).
Somatostatin-receptor positive brain stem glioma visualized by octreoscan. Neuro Endocrinol
Lett 28(3): 250-251.
23) Roujeau T, Machado G, Garnett MR, Miquel C, Puget S, Geoerger B, Grill J, Boddaert
N, Di Rocco F, Zerah M, Sainte-Rose C, (2007). Stereotactic biopsy of diuse pontine lesions
in children. J. Neurosurg 107(1 Suppl): 1-4.
24) Sho A, Kondo S, Kamitani H, Otake H, Watanabe T, (2007). Establishment of
experimental glioma models at the intrinsic brainstem region of the rats. Neurol Res 29(1):
36-42.
25) Siu IM, Tyler BM, Chen JX, Eberhart CG, omale UW, Olivi A, Jallo GI, Riggins
GJ, Gallia GL, (2010). Establishment of a human glioblastoma stemlike brainstem rodent
tumor model. J. Neurosurg Pediatr 6(1): 92-97.
26) omale UW, Tyler B, Renard V, Dorfman B, Chacko VP, Carson BS, Haberl EJ, Jallo
GI, (2009). Neurological grading, survival, MR imaging, and histological evaluation in the
rat brainstem glioma model. Childs Nerv Syst 25(4): 433-441.
27) Whittle IR, Short DM, Deighton RF, Kerr LE, Smith C, McCulloch J, (2007). Proteomic
analysis of gliomas. Br J. Neurosurg 21(6): 576-582.
28) Zarghooni M, Bartels U, Lee E, Buczkowicz P, Morrison A, Huang A, Bouet E,
Hawkins C, (2010). Whole-genome proling of pediatric diuse intrinsic pontine gliomas
highlights platelet-derived growth factor receptor alpha and poly (ADP-ribose) polymerase as
potential therapeutic targets. J. Clin Oncol 28(8): 1337-1344.
29) Zhang S, Feng X, Koga H, Ichikawa T, Abe S, Kumanishi T, (1993). p53 gene mutations
in pontine gliomas of juvenile onset. Biochem Biophys Res Commun 196(2): 851-857.
362
Appendix D: Research Articles
363
Appendix D: Research Articles
References from Chapter 15, Animal Models for DIPG
1) Barth RF, Kaur B, (2009). Rat brain tumor models in experimental neuro-oncology: the
C6, 9L, T9, RG2, F98, BT4C, RT-2 and CNS-1 gliomas. J. Neurooncol 94(3): 299-312.
2) Becher OJ, Hambardzumyan D, Fomchenko EI, Momota H, Mainwaring L, Bleau AM,
et al., (2008). Gli activity correlates with tumor grade in platelet-derived growth factor-induced
gliomas. Cancer Res 68(7): 2241-2249.
3) Becher OJ, Hambardzumyan D, Walker TR, Helmy K, Nazarian J, Albrecht S, Hiner
RL, Gall S, Huse JT, Jabado N, MacDonald TJ, Holland EC, (2010 in press). Preclinical
evaluation of radiation and perifosine in a genetically and histologically accurate model of
brainstem glioma. Cancer Res
4) Brennan C, Momota H, Hambardzumyan D, Ozawa T, Tandon A, Pedraza A, et al.,
(2009). Glioblastoma subclasses can be dened by activity among signal transduction pathways
and associated genomic alterations. PLoS One 4(11):e7752.
5) Hashizume R, Ozawa T, Dinca EB, Banerjee A, Prados MD, James CD, et al., (2010). A
human brainstem glioma xenograft model enabled for bioluminescence imaging. J. Neurooncol
96(2): 151-159.
6) Holland EC, Celestino J, Dai C, Schaefer L, Sawaya RE, Fuller GN., (2000). Combined
activation of Ras and Akt in neural progenitors induces glioblastoma formation in mice. Nat
Genet 25(1): 55-57.
7) Jallo GI, Penno M, Sukay L, Liu JY, Tyler B, Lee J, et al., (2005). Experimental models
of brainstem tumors: development of a neonatal rat model. Childs Nerv Syst 21(5): 399-403.
8) Jallo GI, Volkov A, Wong C, Carson BS, Sr., Penno MB, (2006). A novel brainstem tumor
model: functional and histopathological characterization. Childs Nerv Syst 22(12): 1519-1525.
9) Kondo A, Goldman S, Vanin EF, Sredni ST, Rajaram V, Soares MB, et al., (2009). An
experimental brainstem tumor model using in vivo bioluminescence imaging in rat. Childs
Nerv Syst 25(5): 527-533.
10) Kwon CH, Zhao D, Chen J, Alcantara S, Li Y, Burns DK, et al., (2008). Pten
haploinsuciency accelerates formation of high-grade astrocytomas. Cancer Res 68(9): 3286-
3294.
11) Lee J, Jallo GI, Guarnieri M, Carson BS, Sr., Penno MB, (2005). A novel brainstem
tumor model: guide screw technology with functional, radiological, and histopathological
characterization. Neurosurg Focus 18(6A): E11.
12) Lee J, Kotliarova S, Kotliarov Y, Li A, Su Q, Donin NM, et al., (2006). Tumor stem
cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype
and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell 9(5): 391-403.
13) Liu Q, Liu R, Kashyap MV, Agarwal R, Shi X, Wang CC, et al., (2008). Brainstem
glioma progression in juvenile and adult rats. J Neurosurg 109(5): 849-855.
14) Sho A, Kondo S, Kamitani H, Otake M, Watanabe T, (2007). Establishment of
experimental glioma models at the intrinsic brainstem region of the rats. Neurol Res 29(1):
36-42.
References from Chapter 17, Convection-Enhanced Delivery in DIPG
1) Bobo RH, et al., (1994). Convection-enhanced delivery of macromolecules in the brain.
Proc Natl Acad Sci U S A, 91(6): 2076-2080.
2) Chen MY, et al., (2005). Surface properties, more than size, limiting convective distribution
of virus-sized particles and viruses in the central nervous system. J. Neurosurg 103(2): 311-319.
3) Saito R, et al., (2006). Tissue anity of the infusate aects the distribution volume during
convection-enhanced delivery into rodent brains: implications for local drug delivery. J. Neurosci
Methods 154(1-2): 225-232.
4) Nguyen JB, et al., (2001). Convection-enhanced delivery of AAV-2 combined with heparin
increases TK gene transfer in the rat brain. Neuroreport 12(9): 1961-1964.
5) Cunningham J, et al., (2000). Distribution of AAV-TK following intracranial convection-
enhanced delivery into rats. Cell Transplant 9(5): 585-594.
6) Saito R, et al., (2004). Distribution of liposomes into brain and rat brain tumor models
by convection-enhanced delivery monitored with magnetic resonance imaging. Cancer Res
64(7): 2572-2579.
7) Perlstein B, et al., (2008). Convection-enhanced delivery of maghemite nanoparticles:
Increased ecacy and MRI monitoring. Neuro Oncol 10(2): 153-161.
8) Mardor Y, et al., (2005). Convection-enhanced drug delivery: increased ecacy and magnetic
resonance image monitoring. Cancer Res 65(15): 6858-6863.
9) Varenika V, et al., (2008). Detection of infusate leakage in the brain using real-time imaging
of convection-enhanced delivery. J. Neurosurg 109(5): 874-880.
10) Raghavan R, et al., (2006). Convection-enhanced delivery of therapeutics for brain disease,
and its optimization. Neurosurg Focus 20(4): E12.
11) Oh S, et al., (2007). Improved distribution of small molecules and viral vectors in the
murine brain using a hollow ber catheter. J. Neurosurg 107(3): 568-577.
12) Sampson JH, et al., (2008). Intracerebral infusion of an EGFR-targeted toxin in recurrent
malignant brain tumors. Neuro Oncol 10(3): 320-329.
13) Sampson JH, et al., (2010). Poor drug distribution as a possible explanation for the results
of the PRECISE trial. J. Neurosurg 113(2): 301-309.
14) Sampson JH, et al., (2007). Induction of hyperintense signal on T2-weighted MR images
correlates with infusion distribution from intracerebral convection-enhanced delivery of a tumor-
targeted cytotoxin. AJR Am J Roentgenol 188(3): 703-709.
15) Sampson JH, et al., (2006). Comparison of intratumoral bolus injection and convection-
enhanced delivery of radiolabeled antitenascin monoclonal antibodies. Neurosurg Focus 20(4):
E14.
16) Sampson JH, et al., (2007). Intracerebral infusate distribution by convection-enhanced
delivery in humans with malignant gliomas: descriptive eects of target anatomy and catheter
positioning. Neurosurgery 60(2 Suppl 1): ONS89-98; discussion ONS98-99.
17) Lonser RR, et al., (2007). Real-time image-guided direct convective perfusion of intrinsic
brainstem lesions. Technical note. J. Neurosurg 107(1): 190-197.
18) Heiss JD, et al., (2010). Image-guided convection-enhanced delivery of muscimol to the
primate brain. J. Neurosurg 112(4): 790-795.
19) Croteau D, et al., (2005). Real-time in vivo imaging of the convective distribution of a
low-molecular-weight tracer. J. Neurosurg 102(1): 90-97.
20) Lonser RR, et al., (2002). Successful and safe perfusion of the primate brainstem: in
vivo magnetic resonance imaging of macromolecular distribution during infusion. Neurosurg
364
Appendix D: Research Articles
365
Appendix D: Research Articles
97(4): 905-913.
21) Sampson JH, et al., (2007). Clinical utility of a patient-specic algorithm for simulating
intracerebral drug infusions. Neuro Oncol 9(3): 343-353.
22) Sandberg DI, Edgar MA, Souweidane MM, (2002). Convection-enhanced delivery into
the rat brainstem. J. Neurosurg 96(5): 885-891.
23) Occhiogrosso G, et al., (2003). Prolonged convection-enhanced delivery into the rat
brainstem. Neurosurgery 52(2): 388-393; discussion 393-394.
24) Souweidane MM, et al., (2004). Interstitial infusion of IL13-PE38QQR in the rat brain
stem. J. Neurooncol 67(3): 287-293.
25) Souweidane MM, et al., (2004). Interstitial infusion of carmustine in the rat brain stem
with systemic administration of O6-benzylguanine. J. Neurooncol 67(3): 319-326.
26) Luther N, et al., (2008). Intraparenchymal and intratumoral interstitial infusion of
anti-glioma monoclonal antibody 8H9. Neurosurgery 63(6): 1166-1174; discussion 1174.
27) Luther N, et al., (2010). Interstitial infusion of glioma-targeted recombinant immunotoxin
8H9scFv-PE38. Mol Cancer er 9(4): 1039-1046.
28) Giese H, et al., (2010). Precision of navigated stereotactic probe implantation into the
brainstem. J. Neurosurg Pediatr 5(4): 350-359.
29) Pincus DW, et al., (2006). Brainstem stereotactic biopsy sampling in children. J. Neurosurg
104(2 Suppl): 108-114.
30) Roujeau T, et al., (2007). Stereotactic biopsy of diuse pontine lesions in children. J.
Neurosurg 107(1 Suppl): 1-4.
31) Weber F, et al., (2003). Safety, tolerability, and tumor response of IL4-Pseudomonas exotoxin
(NBI-3001) in patients with recurrent malignant glioma. J. Neurooncol 64(1-2): 125-137.
32) Lidar Z, et al., (2004). Convection-enhanced delivery of paclitaxel for the treatment of
recurrent malignant glioma: a phase I/II clinical study. J. Neurosurg 100(3): 472-479.
33) Krauze MT, et al., (2007). Convection-enhanced delivery of nanoliposomal CPT-11
(irinotecan) and PEGylated liposomal doxorubicin (Doxil) in rodent intracranial brain tumor
xenografts. Neuro Oncol 9(4): 393-403.
34) Debinski W, et al., (1999). Receptor for interleukin 13 is a marker and therapeutic target
for human high-grade gliomas. Clin Cancer Res 5(5): 985-990.
35) Debinski W, et al., (1999). Receptor for interleukin 13 is abundantly and specically over-
expressed in patients with glioblastoma multiforme. Int J. Oncol 15(3): 481-486.
36) Johnson VG, et al., (1988). e role of the diphtheria toxin receptor in cytosol translocation.
J. Biol Chem 263(3): 1295-1300.
37) Weaver M, Laske DW, (2003). Transferrin receptor ligand-targeted toxin conjugate (Tf-
CRM107) for therapy of malignant gliomas. J. Neurooncol 65(1): 3-13.
38) Torp SH, et al., (1991). Epidermal growth factor receptor expression in human gliomas.
Cancer Immunol Immunother 33(1): 61-64.
39) Debinski W, et al., (1995). A novel chimeric protein composed of interleukin 13 and
Pseudomonas exotoxin is highly cytotoxic to human carcinoma cells expressing receptors for
interleukin 13 and interleukin 4. J. Biol Chem 270(28): 16775-16780.
40) Kunwar S, et al., (2007). Direct intracerebral delivery of cintredekin besudotox
(IL13-PE38QQR) in recurrent malignant glioma: a report by the Cintredekin Besudotox
Intraparenchymal Study Group. J. Clin Oncol 25(7): 837-844.
41) Liu H, et al., (2000). Interleukin-13 sensitivity and receptor phenotypes of human glial
cell lines: non-neoplastic glia and low-grade astrocytoma dier from malignant glioma. Cancer
Immunol Immunother 49(6): 319-324.
42) Joshi BH, Plautz GE, Puri RK, (2000). Interleukin-13 receptor alpha chain: a novel
tumor-associated transmembrane protein in primary explants of human malignant gliomas.
Cancer Res 60(5): 1168-1172.
43) Madhankumar AB, Mintz A, Debinski W, (2004). Interleukin 13 mutants of enhanced
avidity toward the glioma-associated receptor, IL13Ralpha2. Neoplasia 6(1): 15-22.
44) Patel SJ, et al., (2005). Safety and feasibility of convection-enhanced delivery of Cotara
for the treatment of malignant glioma: initial experience in 51 patients. Neurosurgery 56(6):
1243-1252; discussion 1252-1253.
45) Verel I, Visser GW, van Dongen GA, (2005). The promise of immuno-PET in
radioimmunotherapy. J. Nucl Med 46 Suppl 1: 164S-1671S.
46) Becher OJ, et al., (2010). Preclinical evaluation of radiation and perifosine in a genetically
and histologically accurate model of brainstem glioma. Cancer Res 70(6): 2548-2557.
47) Zarghooni M, et al., (2010). Whole-genome proling of pediatric diuse intrinsic pontine
gliomas highlights platelet-derived growth factor receptor alpha and poly (ADP-ribose) polymerase
as potential therapeutic targets. J. Clin Oncol 28(8): 1337-1344.
48) Paugh BS, et al., (2010). Integrated molecular genetic proling of pediatric high-grade
gliomas reveals key dierences with the adult disease. J. Clin Oncol 28(18): 3061-3068.
49) Gilbertson RJ, et al., (2003). ERBB1 is amplied and overexpressed in high-grade diusely
inltrative pediatric brain stem glioma. Clin Cancer Res 9(10 Pt 1): 3620-3624.
50) Joshi BH, et al., (2008). Identification of interleukin-13 receptor alpha2 chain
overexpression in situ in high-grade diusely inltrative pediatric brainstem glioma. Neuro
Oncol 10(3): 265-274.
51) Parsons DW, et al., (2008). An integrated genomic analysis of human glioblastoma
multiforme. Science 321(5897): 1807-1812.
References from Chapter 19, DIPG and Tissue Donation
1) Albright AL, Packer RJ, Zimmerman R, Rorke LB, Boyett J, Hammond GD., (Dec
1993). Magnetic resonance scans should replace biopsies for the diagnosis of diuse brain
stem gliomas: a report from the Children's Cancer Group. Neurosurgery 33(6): 1026-1029;
discussion 1029-1030.
2) Broniscer A, Baker JN, Baker SJ, Chi SN, Geyer JR, Morris EB, Gajjar A., (Oct 2010).
Prospective collection of tissue samples at autopsy in children with diuse intrinsic pontine
glioma. Cancer 116(19): 4632-4637.
3) Angelini P, Hawkins C, Laperriere N, Bouet E, Bartels U., (Jan 2011). Post mortem
examinations in diuse intrinsic pontine glioma: challenges and chances. J. Neurooncol
101(1): 75-81
4) Zarghooni M, Bartels U, Lee E, Buczkowicz P, Morrison A, Huang A, Bouet E, Hawkins
C., (Mar 10, 2010). Whole-genome proling of pediatric diuse intrinsic pontine gliomas
highlights platelet-derived growth factor receptor alpha and poly (ADP-ribose) polymerase as
potential therapeutic targets. J. Clin Oncol 28(8): 1337-1344.
366
Appendix D: Research Articles
367
Appendix D: Research Articles
5) Paugh BS, Broniscer A, Qu C, Miller CP, Zhang J, Tatevossian RG, Olson JM, Geyer JR,
Chi SN, da Silva NS, Onar-omas A, Baker JN, Gajjar A, Ellison DW, Baker SJ., (Oct 20,
2011). Genome-wide analyses identify recurrent amplications of receptor tyrosine kinases and
cell-cycle regulatory genes in diuse intrinsic pontine glioma. J. Clin Oncol 29(30): 3999-4006.
6) Puget S, Philippe C, Bax DA, Job B, Varlet P, Junier MP, Andreiuolo F, Carvalho D, Reis
R, Guerrini-Rousseau L, Roujeau T, Dessen P, Richon C, Lazar V, Le Teu G, Sainte-Rose
C, Geoerger B, Vassal G, Jones C, Grill J., (2012). Mesenchymal transition and PDGFRA
amplication/mutation are key distinct oncogenic events in pediatric diuse intrinsic pontine
gliomas. PLoS One 7(2): 303-313.
7) Roujeau T, Machado G, Garnett MR, Miquel C, Puget S, Geoerger B, Grill J, Boddaert
N, Di Rocco F, Zerah M, Sainte-Rose C., (Jul 2007). Stereotactic biopsy of diuse pontine
lesions in children. J. Neurosurg 107(1 Suppl): 1-4.
References from Chapter 20, Organ and Tissue Donation
1) Armanios A, Grossman S, Yang S, et al. (2004). Transmission of glioblastoma multiforme
following bilateral lung transplantation from an aected donor: Case study and review of the
literature. Neuro-oncology 6(3): 259-263.
2) Berger M, Baumeister B, Geyer J, Milstein J, Kaney P, LeRoux P, (1991). The risks
of metastases from shunting in children with primary central nervous system tumors. J.
Neurosurgery 74: 872-877.
3) Buell J, Trofe J, Sethuraman G, et al. (2003). Donors with central nervous system
malignancies: Are they truly safe? Transplantation 76(2): 340-343.
4) Buell J, Grossa T, Allowaya RR, Trofea J, Woodle ES, (2005). Central nervous system
tumors in donors: Misdiagnosis carries a high morbidity and mortality. Transplantation
Proceedings 37(2): 583-584.
5) Campbell A, Chan H, Becker L, Daneman A, Park T, Homan H, (1984). Extracranial
metastases in childhood primary intracranial tumors: A report of 21 Cases and review of the
Literature. Cancer 53: 974-981.
6) Cavaliere R, Schi D, (2004). Donor transmission of primary brain tumors: A neurooncologic
perspective. Transplantation Review 18(4): 204-213.
7) Chui AK, Herbertt K, Wang LS, et al., (1999). Risk of tumor transmission in
transplantation from donors with primary brain tumors: An Australian and New Zealand
registry report. Transplantation Proceedings 31: 1266-1267.
8) Collignon FP, Holland EC, Feng S, (2004). Organ donors with malignant gliomas: an
update. Am J.Transplantation 4: 15-21.
9) Colquhoun SD, Rober ME, Shaked A, (1994). Transmission of CNSmalignancy by organ
transplantation. Transplantation 57: 970-974.
10) Council of Europe, (1997). International Consensus Document: Select committee of experts
on the organizational aspects of co-operation in organ transplantation. Standardization of organ
donor screening to prevent transmission of neoplastic disease. Transplant Newsletter 2: 4-10.
11) Halpern SD, Shaked A, Hasz RD,Caplan AL, (2008). Informing candidates for solid-
organ transplantation about donor risk factors. New Eng J.Med 358: 2832-2837.
12) Homan H, Duner P, (1985). Extraneural metastases of central nervous system tumors.
Cancer 56: 1778-1782
13) Hornick L, Tenderich G, Wlost S, Zittermann A, Minami K, Koerfer R, (2004).
Organs from donors with primary brain malignancy: e fate of cardiac allograft recipients.
Transplantation Proceedings 36: 3133-3137.
14) Jonas S, Bechstein WO, Lemmens H-P, et al., (1996). Liver graft-transmitted glioblastoma
multiforme. A case report and experience with 13 multiorgan donors suering from primary
cerebral neoplasia. Transplantation International 9: 426-429.
15) Kashyap R, Ryan C, Sharma R, et al., (2009). Liver grafts from donors with central
nervous system tumors: A single-centre perspective. Liver Transplantation 15: 1204-1208.
16) Kauman H, McBride M, Cherikh W, Spain P, Marks W, Roza A, (2002). Transplant
tumor registry: Donor related malignancies. Transplantation 74: 358-362.
17) Kauman HM, (2005). e United Network for Organ Sharing position on using donors
with primary central nervous system malignancies. Transplantation 79: 622-623.
18) Kauman HM, Cherikh WS, McBride MA, Cheng Y, Hanto DW, (2007). Deceased
donors with a past history of malignancy: An Organ Procurement and Transplantation Network/
United Network for Organ Sharing update. Transplantation 84: 272-274.
19) Kleinman G, Hochberg F, Richardson E, (1981). Systemic metastases from
medulloblastoma. Cancer 48: 2296-2309.
20) Newton HB, Henson J, Walker RW, (1992). Extraneural metastases in ependymoma. J.
Neuro Onc 14: 135-142.
21) OPTN Organ Procurement and Transplantation Network Annual Report, (2009).
Accessed Jun 1, 2011: http://optn.transplant.hrsa.gov/ar2009/default.htm.
22) Pasquier B, Pasquier D, N’Golet A, (1980). Extraneural metastases of astrocytomas
and glioblastomas: Clinicopathological study of two cases and review of literature. Cancer 45:
112-125.
23) Penn I, (1997).Transmission of cancer from organ donors. Annals of Transplantation 2:
7-12.
24) Pokorna E, Vitko S, (2001). e fate of recipients of organs from donors with diagnosis
of primary brain tumor. Transplant Int 14: 346-347.
25) Punnett AS, McCarthy L, Dirks P, Hawkins C, Bouet E, (2004). Patients with primary
brain tumors as organ donors: Case report and review of the literature. Pediatric Blood &
Cancer 43(1):73-77.
26) Schweitzer T, Vince GH, Herbold C, Roosen K, Tonn JC, (2001). Extraneural metastases
of primary brain tumors. J. Neuro-Oncology 53: 107-114.
27) Subramanian A, Harris A, Piggott K, Schi C, Bradford R, (2002). Metastasis to and
from the central nervous system--the ‘relatively protected site.’ Lancet Oncology 3: 498-507.
28) Watson CJE, Roberts R, Wright KA, (2010). How safe is it to transplant organs from
deceased donors with primary intracranial malignancy? An analysis of UK registry data. Am
J. Transplantation 10: 1437-1444.
References from Chapter 22, Journey of Sadness and Hopes
1) Feudtner C., (Dec 10 2009). e breadth of hopes. New Eng J. Med. 361(24): 2306-2307.
2) Feudtner C., (2005). Hope and the prospects of healing at the end of life. J. Altern
Complement Med. 11 Suppl 1: S23-30.
3) Hinds PS, Oakes LL, Hicks J, et al., (Dec 10 2009). "Trying to be a good parent" as
368
Appendix D: Research Articles
dened by interviews with parents who made phase I, terminal care, and resuscitation decisions
for their children. J. Clin Oncol. 27(35): 5979-5985.
4) Duggleby W, Holtslander L, Steeves M, Duggleby-Wenzel S, Cunningham S., (Sep
2010). Discursive meaning of hope for older persons with advanced cancer and their caregivers.
Can J. Aging. 29(3): 361-367.
5) Mack JW, Hilden JM, Watterson J, et al., (Dec 20 2005). Parent and physician perspectives
on quality of care at the end of life in children with cancer. J. Clin Oncol. 23(36): 9155-9161.
6) Mack JW, Weeks JC, Wright AA, Block SD, Prigerson HG., (Mar 1 2010). End-of-life
discussions, goal attainment, and distress at the end of life: predictors and outcomes of receipt of
care consistent with preferences. J. Clin Oncol. 28(7): 1203-1208.
7) Meyer EC, Ritholz MD, Burns JP, Truog RD., (Mar 2006). Improving the quality of
end-of-life care in the pediatric intensive care unit: parents' priorities and recommendations.
Pediatrics. 117(3): 649-657.

